Les informations pratiques concernant le déroulement de l’unité d’enseignement RCP209 « Apprentissage, réseaux de neurones et modèles graphiques » au Cnam se trouvent dans ce préambule.
« Prediction can be very difficult, especially about the future. »
(Niels Bohr, cité dans Teaching and Learning Elementary Social Studies, Arthur K. Ellis, 1970, p. 431.)
Quelle sera la quantité d’algues vertes à ramasser durant le mois de juillet prochain dans la baie de Saint-Brieuc ? Quel est l’âge d’un Fagus sylvatica (hêtre) qui a une circonférence de 236 cm ? Quel chiffre représente l’image ?
Dans chacun de ces exemples, nous connaissons les valeurs de certaines variables « explicatives » et souhaitons obtenir la valeur d’une variable « expliquée » (ou « à prédire ») qui doit dépendre, de façon significative, des variables explicatives. Si on considère le premier exemple, la variable expliquée correspond à la quantité d’algues vertes échouées dans la baie de Saint-Brieuc sur une période bien définie et l’ensemble des variables explicatives inclut le volume d’épandages dans les bassins hydrographiques environnants lors des 12 mois qui précèdent, la quantité de pluie, l’ensoleillement, etc.
Afin de répondre aussi précisément que possible à la question posée il est nécessaire d’identifier les variables explicatives et de modéliser les dépendances entre celles-ci et la variable expliquée.
L’identification de variables explicatives pour un problème donné devrait être basée sur une compréhension au moins partielle du phénomène concerné. De plus en plus souvent, cette identification passe plutôt par la sélection, explicite ou non, de variables dans un grand ensemble de variables dont les valeurs sont accessibles. Nous reviendrons plus tard sur ce processus et sur les différents risques liés à la recherche dans un trop grand ensemble de variables candidates.
Un modèle des dépendances entre les variables doit permettre d’obtenir une estimation pour la valeur de la variable expliquée à partir des valeurs prises par les variables explicatives à un moment donné (ces valeurs forment une « observation »). Nous parlons de modèle décisionnel car la valeur estimée constitue le support d’une décision. On emploie également parfois les termes « prédiction » et « modèle prédictif » (même si aucun aspect temporel n’est présent dans la dépendance modélisée) car le modèle est obtenu à partir d’observations passées et peut servir pour de nouvelles observations.
Un modèle décisionnel peut être construit analytiquement, grâce à une parfaite compréhension du phénomène observé, ou par des moyens statistiques, sur la base d’un ensemble d’observations disponibles. Dans ce cours nous examinons la modélisation décisionnelle (ou prédictive) à partir de données. Par données nous entendons ici des observations caractérisées par les valeurs prises par un ensemble de variables. La modélisation est décisionnelle car elle vise la capacité à prédire, pour chaque nouvelle observation, la valeur (inconnue) d’une variable expliquée à partir des valeurs (connues) des variables explicatives. La modélisation est à partir de données car nous nous intéressons à la construction en grande partie automatique de modèles sur la base des observations disponibles.
Il faut toutefois remarquer que la construction préalable d’un modèle n’est pas indispensable. Ainsi, la valeur de la variable expliquée peut être estimée pour une nouvelle observation à partir, par exemple, des k plus proches observations pour lesquelles la valeur de la variable expliquée est connue.
La modélisation décisionnelle trouve ses nombreuses applications à la fois dans des problèmes de « reconnaissance des formes » et dans des problèmes de « fouille de données ».
La reconnaissance des formes (pattern recognition) consiste, au sens strict, à identifier à quelle catégorie appartient une « forme » décrite par des données brutes. Trouver quel chiffre représente l’image est un problème de classement qui fait partie de la reconnaisance des formes au sens strict. Au sens large, la reconnaissance des formes inclut les problèmes de régression correspondant aux deux autres questions mentionnées au début de cette section.
La fouille de données (data mining) consiste, au sens strict, à rechercher des régularités ou des relations inconnues a priori dans de grands volumes de données. Si cette définition met plutôt en évidence un objectif descriptif, le but ultime de la fouille de données est en général d’obtenir des modèles décisionnels permettant d’identifier les données « intéressantes » et de prendre des décisions pour ces données.
Lorsqu’on s’intéresse à la modélisation décisionnelle à partir de données, il est important de comprendre d’abord la nature des problèmes de modélisation. Lors de la séance introductive nous examinerons d’abord les différents types de problèmes : le classement (la discrimination), la régression et la prédiction structurée. Un modèle est construit à partir d’un ensemble d’observations disponibles (pour lesquelles les valeurs prises par toutes les variables, y compris la variable expliquée, sont connues) et doit être utilisé ensuite pour de nouvelles observations afin d’estimer les valeurs prises par la variable expliquée. La capacité d’un modèle à généraliser, c’est à dire à faire de bonnes prédictions pour de nouvelles observations, est critique. Nous nous intéresserons donc aux moyens disponibles pour estimer la capacité de généralisation d’un modèle. Enfin, nous regarderons la méthodologie générale de construction, d’évaluation et de sélection de modèles décisionnels.
La suite du cours vise à vous permettre de maîtriser plusieurs outils de modélisation décisionnelle parmi les plus utilisés aujourd’hui, notamment les forêts d’arbres de décision, les machines à vecteurs support (support vector machines, SVM) et les réseaux de neurones profonds (deep networks).
Les travaux pratiques (TP) sont une mise en œuvre directe de notions et méthodes vues en cours. Les premières séances s’intéressent à de la méthodologie de construction, évaluation et sélection de modèles décisionnels. Les séances suivantes permettent de mettre en œuvre les arbres de décision et les forêts d’arbres de décision, les machines à vecteurs support et les réseaux de neurones profonds. L’outil employé pour les TP est Scikit-learn, libre et ouvert, utilisant le langage Python. Si vous avez suivi l’UE RCP208 vous avez déjà eu l’occasion de vous en servir. Pour les TP concernant l’apprentissage profond nous nous servons de Keras.
De nombreuses références, sous forme imprimée ou sur le Web, traitent les différents sujets abordés dans cet enseignement. Des références spécifiques vous seront également indiquées dans les séances de cours et de TP. Nous avons listé ci-dessous des ouvrages qui couvrent des parties plus larges du contenu de cet enseignement.
Dreyfus, G., J. Martinez, M. Samuelides, M. Gordon, F. Badran, S. Thiria. Apprentissage statistique : Réseaux de neurones - Cartes topologiques - Machines à vecteurs supports. Éditions Eyrolles, 2008, 3ème édition.
Schölkopf, B., A. Smola. Learning with Kernels. MIT Press, 2002.
Chaque problème traité, par exemple celui de la prédiction de la quantité d’algues vertes à ramasser, est décrit par un ensemble de variables « explicatives » et une variable à expliquer. Afin de construire un modèle décisionnel à partir de données, il est nécessaire de disposer d’un certain nombre d”observations concernant toutes les variables considérées. Pour une variable aléatoire simple, une observation correspond à la valeur de la variable à un moment donné. Dans le cas d’un problème de modélisation caractérisé par plusieurs variables, une observation inclut en général les valeurs prises à un même moment par toutes les variables. Par exemple, pour l’identification du chiffre représenté par une image on considère les valeurs prises par les pixels de l’image (donc par une partie d’un champ de vision à un moment donné).
Les données utilisées pour la construction de modèles sont des ensembles d’observations qui doivent inclure, pour au moins une partie d’entre elles, à la fois les valeurs des variables explicatives et celles correspondantes de la variable expliquée. Nous pouvons parfois nous trouver en présence de quelques observations incomplètes, ou à données manquantes.
Un modèle décisionnel sera validé sur des observations de même type, comportant les valeurs des variables explicatives et celles associées de la variable expliquée. Une fois validé, ce modèle sera en revanche employé pour des observations qui se limitent aux valeurs des variables explicatives afin d’obtenir une estimation pour la valeur correspondante (inconnue) de la variable expliquée.
Les principales différences entre problèmes de décision sont issues de la nature des variables explicatives. Pour mieux comprendre ces différences, il est utile de considérer les exemples suivants :
Quel chiffre représente l’image ?
Une région d’une image représente un visage ou non ?
Les symptômes correspondent à la maladie A ou B ou C ou aucune ?
Quelle sera la quantité d’algues vertes à ramasser durant le mois de juillet prochain dans la baie de Saint-Brieuc ?
Quel sera le débit de la Loire à Tours dans 48h ?
Quel est l’âge d’un Fagus sylvatica (hêtre) qui a une circonférence de 236 cm ?
Quelle est l’entité nommée dans « La Maison Blanche a démenti ces informations. » ?
Quelle est la région d’image correspondant aux pantalons ?
Cela nous permet de mettre en évidence trois types de problèmes de décision :
Classement (ou discrimination, classification en anglais) : la variable expliquée est une variable nominale (variable à modalités), chaque observation est associée à une modalité et une seule (appelée en général classe). Les exemples 1, 2 et 3 ci-dessus rentrent dans cette catégorie. Pour le premier exemple, la variable expliquée possède 10 modalités qui correspondent aux chiffres de 0 à 9 (éventuellement 11 modalités si on considère une classe « autre »). Pour le deuxième, la variable expliquée a deux modalités, « visage » et « non visage ». Dans le troisième exemple, la variable expliquée possède quatre modalités : A, B, C et « autre ».
Régression (regression) : la variable expliquée est une variable quantitative qui prend des valeurs dans un sous-domaine de l’ensemble \(\mathbb{R}\) des rééls. Les exemples 4, 5 et 6 font partie de cette catégorie, les variables expliquées étant respectivement la quantité, le débit et l’âge.
Prédiction structurée (structured prediction) : la variable expliquée prend des valeurs dans un domaine de données structurées. Les exemples 7 et 8 ci-dessus rentrent dans cette catégorie. Dans l’exemple 7, les valeurs que peut prendre la variable expliquée sont les parties (les sous-séquences de mots) de la phrase. Dans l’exemple 8, les valeurs possibles de la variable expliquée sont les sous-ensembles de pixels de l’image.
Dans le contexte de la modélisation décisionnelle, par modèle nous entendons en général une règle de décision.
Si nous prenons le cas d’un problème de classement ou de discrimination entre deux classes, la règle de décision consistera d’abord en une frontière de discrimination. La figure suivante présente un ensemble d’observations bidimensionnelles (les points rouges et les points verts), les variables explicatives étant dans ce cas l’abscisse \(x\) et l’ordonnée \(y\) de chaque point. Pour chaque observation, la valeur de la variable nominale expliquée est soir « rouge » soit « vert ». Exprimé autrement, chaque observation appartient soit à la classe « rouge » soit à la classe « verte ». Le trait bleu foncé est une frontière de discrimination qui permet de séparer (de manière imparfaite) les points rouges des points verts. Une nouvelle observation qui est située d’un côté de cette frontière est « affectée » à la classe correspondante (la valeur corespondante est prédite par le modèle pour la variable nominale expliquée).
Fig. 1 Exemple de frontière de discrimination pour problème de classement à 2 classes¶
Le modèle peut éventuellement être complété par des critères de rejet (ou de refus d’affectation) qui donnent la possibilité de refuser de décider pour certaines observations. Il faut noter que, d’un point de vue pratique, le rejet peut ne pas être autorisé ou alors est associé à une valeur spécifique de la variable expliquée (par ex. classe « autre » pour un problème de classement). Par ailleurs, obtenir de bons critères de rejet pour un problème particulier soulève des difficultés spécifiques par rapport à la seule estimation de la valeur de la variable expliquée (nominale, quantitative ou structurée).
Il est d’abord envisageable de refuser de prendre une décision de classement pour les observations qui sont « trop » proches de la frontière de discrimination, comme dans la figure suivante. On parle alors de rejet d’ambiguïté car l’incertitude peut être élevée quant à la position exacte de la frontière ou alors les classes se superposent partiellement dans le voisinage de cette frontière.
Fig. 2 Exemple de zone de rejet d’ambiguïté (en jaune)¶
Enfin, il est envisageable de refuser de classer les observations qui sont « trop » éloignées des observations connues, comme dans la figure suivante. On parle dans ce cas de rejet de non représentativité, le modèle étant considéré peu fiable aussi loin de la majeure partie des observations.
Fig. 3 Exemple de zone de rejet de non représentativité (en jaune)¶
Sur les mêmes données, des modèles différents peuvent faire des prédictions différentes. Pour un problème de classement à deux classes, la figure suivante présente trois modèles décisionnels (reduits ici à la frontière de discrimination, sans rejet) appliqués à un même ensemble d’observations bidimensionnelles. Le premier modèle correspond à une frontière simple, linéaire, et classe mal (du mauvais côté) un nombre relativement élevé d’observations. Le second correspond à une frontière plus complexe, non linéaire (mais plutôt lisse) et fait comparativement moins d’erreurs de classement. Le troisième modèle correspond à une frontière non linéaire plus complexe (présente des parties avec une courbure plus forte). Nous verrons plus loin comment choisir le « bon » modèle et quels sont les liens entre la complexité des modèles et leurs erreurs.
Fig. 4 Exemple de trois frontières de discrimination pour les mêmes données¶
Considérons maintenant un problème de régression qui consiste à estimer les valeurs d’une variable expliquée \(Y\) (en ordonnée) à partir des valeurs d’une variable explicative \(X\) (en abscisse). La figure suivante présente trois modèles décisionnels (dans ce cas, des règles de prédiction représentées par le trait noir) appliqués à un même ensemble d’observations. Le premier modèle correspond à une prédiction linéaire de type \(y = a x + b\), avec \(a\) et \(b\) constantes. Les deux autres modèles correspondent à des règles de prédiction non linéaires.
Fig. 5 Exemple de trois règles de prédiction pour les mêmes données¶
Dans le cas de la prédiction structurée, un modèle correspond aussi à une règle de prédiction. Illustrer une telle règle pour un ensemble d’observations est plus ardu, nous nous contenterons ici d’indiquer le résultat dans deux exemples différents mais pour une seule observation à chaque fois.
Le premier exemple concerne la détection d’entités nommées dans des phrases. Pour l’observation (la phrase) « La Maison Blanche a démenti ces informations. », le résultat (la prédiction) du modèle peut être, par exemple, La Maison Blanche. A première vue, il devrait être possible d’aborder un tel problème comme un problème de classement des mots individuels, faisant partie ou non de l’entité nommée. Mais cette approche ignorerait les fortes dépendances entre les classements des différents mots composant une même entité nommée (l’inclusion de « Maison » dans l’entité nommée dépend de l’inclusion de « Blanche » et aussi de l’inclusion de « La »). Par ailleurs, une même phrase peut contenir plusieurs entités nommées (par ex. « La Maison Blanche et Downing Street […] ») et leur nombre n’est pas connu a priori.
Comme second exemple de prédiction structurée nous considérons la détection avec segmentation de vêtements dans des images. Pour l’observation (image) suivante à gauche, le résultat du modèle peut être, par exemple, celui illustré dans l’image de droite (voir [YRCF17]). Les pantalons de l’image de gauche, par exemple, correspondent à la région bleu clair de l’image de droite. Ici encore, nous pouvons éventuellement envisager d’aborder ce problème comme un problème de classement de pixels individuels en autant de classes qu’il y a de catégories de vêtements (plus une classe « autre »). Une telle approche ignorerait les dépendances entre les affectations des différents pixels composant la région représentant un vêtement, ainsi que les dépendances entre les décisions concernant des vêtements différents.
Fig. 6 Exemple de détection et segmentation de vêtements¶
Nous avons mentionné plus tôt la possibilité de construire analytiquement un modèle décisionnel grâce à une parfaite compréhension du phénomène observé. De tels modèles sont très courants. Des exemples simples sont le calcul du temps de vol d’un mobile à partir de la distance à parcourir et de la vitesse, ou le calcul de la concentration d’un produit d’une réaction chimique à partir des concentrations de réactifs et de la température. Les modèles décisionnels sont construits, dans de tels cas, à partir d’une connaissance de lois en physique ou en chimie. Toutefois, ces modèles négligent l’impact d’un ensemble de phénomènes ou de variables non contrôlables. Par exemple, le déplacement des masses d’air a des conséquences sur la vitesse (et sur la trajectoire exacte) d’un avion, donc sur le temps de vol. Le caractère inhomogène de la température ou de la concentration de réactifs a un impact sur la concentration du produit de la réaction chimique. L’exactitude des prédictions de ces modèles est donc variable.
La construction de modèles à partir de données, par des moyens statistiques, sur la base d’un ensemble d’observations disponibles, permet d’aborder les situations dans lesquelles la compréhension des phénomènes (physiques, chimiques, économiques, sociologiques, etc.) impliqués est insuffisante et un modèle analytique ne peut fournir de prédictions de qualité raisonnable. Par ailleurs, la modélisation à partir de données peut parfois contribuer à corriger les prédictions de modèles analytiques qui couvrent le problème de façon incomplète, comme ceux mentionnés plus haut.
Pour la modélisation à partir de données, une première approche est l”apprentissage supervisé qui exploite exclusivement les données (les observations) pour lesquelles sont connues non seulement les valeurs des variables explicatives mais aussi celles correspondantes de la variable expliquée. Ces valeurs de la variable expliquée constituent l’information de « supervision ». L’illustration suivante montre ces observations pour le problème de classement à deux classes que nous avons considéré plus haut.
Fig. 7 Apprentissage supervisé : utilisation exclusive des observations pour lesquelles les valeurs de la variable expliquée sont connues¶
L’information de supervision (les valeurs de la variable expliquée) est souvent difficile à obtenir. Par exemple, pour le problème de classement de chiffres manuscrits, il est nécessaire qu’un humain réalise ce classement. Pour la détection et segmentation de vêtements, un humain doit délimiter les régions correspondantes de chaque image et les affecter à une catégorie. Ces opérations sont coûteuses. Pour déterminer de façon fiable l’âge d’un hêtre qui a une certaine circonférence il est nécessaire de le couper (pour compter le nombre d’anneaux de croissance).
Par apprentissage supervisé il faut donc souvent construire un modèle à partir d’un assez faible nombre d’observations (celles qui possèdent l’information de supervision). En revanche, obtenir un grand nombre d’observations avec des valeurs pour les seules variables explicatives est bien plus facile. Est-ce envisageable d’employer ces nombreuses observations sans supervision afin d’améliorer la qualité d’un modèle par rapport à l’utilisation exclusive des observations avec supervision ? La réponse peut être positive et cette démarche s’inscrit dans l”apprentissage semi-supervisé (voir par ex. [CScholkopfZ06]). L’apprentissage semi-supervisé considère que l’information de supervision est disponible sous une forme partielle, par ex. pour une (faible) partie des données (observations). Par ailleurs, des connaissances a priori concernant le problème traité permettent éventuellement de valider des hypothèses sur le lien entre la distribution des données et la variable expliquée. Une telle hypothèse est, par ex., que la frontière de séparation entre classes se trouve dans une région de faible densité, comme dans la figure suivante. Ce sont ces hypothèses qui permettent de se servir des données sans information de supervision afin d’améliorer la modélisation décisionnelle.
Fig. 8 Apprentissage semi-supervisé : tient compte non seulement des observations avec supervision (points pleins dans cette figure), mais également de celles sans supervision (points creux dans la figure)¶
La construction de modèles décisionnels à partir de données exploite un ensemble d’observations pour lesquelles l’information de supervision est présente (les valeurs de la variable expliquée sont connues). Une fois construits, ces modèles sont employés pour faire des prédictions, c’est à dire estimer les valeurs de la variable expliquée, sur de nouvelles observations pour lesquelles l’information de supervision est absente. Par capacité de généralisation d’un modèle on entend la capacité du modèle à faire de bonnes prédictions sur ces nouvelles observations.
Afin de construire un modèle décisionnel il est en général nécessaire de
Choisir une famille paramétrique dans laquelle sera recherché le modèle, et ensuite
Optimiser les paramètres afin de trouver le « meilleur » modèle dans cette famille.
Pour illustrer cela, revenons au problème de classement à deux classes considéré plus haut. La figure suivante présente l’ensemble d’observations bidimensionnelles (les points rouges et les points verts) pour lesquelles l’information de supervision (classe « rouge » ou « verte ») est connue. Avec ces observations nous cherchons à déterminer un modèle qui est une frontière de discrimination entre les deux classes.
Nous pouvons d’abord examiner la famille paramétrique des frontières linéaires, représentées par des droites dans le plan des observations bidimensionnelles. Une fois la famille choisie, la recherche d’un modèle se fait par une méthode qui optimise un critère particulier. Si la méthode employée est l”analyse factorielle discriminante (AFD), nous obtenons dans cet exemple la frontière linéaire indiquée dans la figure suivante.
Sur l’ensemble d’observations ayant servi à obtenir le modèle (données d”apprentissage), la qualité de ce modèle est en général évaluée à travers son erreur d’apprentissage (ou risque empirique). Dans cet exemple, elle est mesurée par le pourcentage d’erreurs de classement (observations mal classées). Pour le modèle linéaire obtenu par AFD le risque empirique est ici de 12%.
Considérons maintenant la famille définie par les perceptrons multi-couche (PMC) à une couche cachée de 10 neurones à fonction d’activation \(tanh\) (tangente hyperbolique). Les paramètres à optimiser sont, dans ce cas, les poids des connexions et les seuils des neurones.
La recherche des paramètres optimaux, visant ici à minimiser la somme entre l’erreur d’apprentissage et un terme d”« oubli » avec une pondération \(\alpha = 10^{-5}\), donne comme résultat la frontière non linéaire indiquée dans la figure suivante. Pour ce modèle non linéaire le risque empirique est de seulement 2,3%, bien pus faible que pour le modèle linéaire obtenu par AFD.
Fig. 11 Frontière non linéaire obtenue par perceptron multi-couche (PMC) avec \(\alpha = 10^{-5}\). Erreur d’apprentissage = 2,3%¶
Les modèles obtenus doivent ensuite être employés pour estimer les valeurs de la variable expliquée sur de nouvelles observations pour lesquelles l’information de supervision est absente. L’erreur qui nous intéresse pour l’utilisation d’un modèle n’est donc pas l’erreur d’apprentissage mais plutôt l”erreur de généralisation (ou risque espéré).
Pour le même problème de classement, considérons un autre ensemble d’observations qui possèdent l’information de supervision, qui est issu de la même distribution que les données d’apprentissage mais qui présente une intersection vide avec ces données. La figure suivante montre un tel ensemble. Les observations sont représentées par des points creux, la couleur de chaque point correspond à la classe à laquelle il appartient rééllement. Une erreur de classement est comptée chaque fois qu’une observation se trouve du mauvais côté de la frontière de discrimination (ici, de l’autre côté que la majorité des observations de la même classe).
Fig. 12 Données de test (non utilisées pour l’apprentissage)¶
Nous pouvons tester chaque modèle obtenu plus haut sur ce nouvel ensemble d’observations, que nous appellerons données de test. Si ces observations sont suffisamment nombreuses et issues de la même distribution que les observations futures alors l’erreur d’un modèle sur ces données de test est une bonne indication de son erreur de généralisation (ou risque espéré). Les deux figures suivantes permettent de visualiser les décisions des deux modèles trouvés plus haut (modèle linéaire obtenu par AFD et respectivement modèle non linéaire PMC). Rappelons que la couleur de chaque point correspond à la classe à laquelle il appartient rééllement.
Fig. 13 Classement avec la frontière linéaire obtenue par analyse factorielle discriminante (AFD). Erreur de test = 14%¶
Fig. 14 Classement avec la frontière non linéaire obtenue par perceptron multi-couche (PMC) avec \(\alpha = 10^{-5}\). Erreur de test = 6%¶
Dans cet exemple nous constatons que l’erreur de test est bien plus faible pour ce modèle non linéaire que pour le modèle linéaire. Si l’erreur de test est une bonne indication du risque espéré, entre ces deux modèles nous préférerons nous servir du modèle non linéaire trouvé. Mais comment savoir si nous avons identifié le modèle qui présente la meilleure généralisation dans une catégorie de modèles « accessibles » (que nous pouvons déterminer avec les données, les éventuelles connaissances a priori et les outils disponibles) ? Au sein d’une famille de modèles, comment choisir le critère à optimiser ? Quel est le lien entre le risque espéré (l’erreur de généralisation) et le risque empirique (l’erreur d’apprentissage) ? Nous examinerons ces questions dans la suite.
Nous avons vu que, pour construire un modèle décisionnel, il était nécessaire de choisir une ou plusieurs familles paramétriques et, dans chaque famille, déterminer les paramètres qui définissent le meilleur modèle en optimisant un critère. Avec les seules données d’apprentissage, le seul critère accessible est l’erreur d’apprentissage. Or, c’est l’erreur de généralisation qui nous intéresse mais elle ne peut être directement mesurée car les données futures sont inconnues (ou, du moins, l’information de supervision les concernant) au moment où le modèle est mis au point. Comment obtenir le modèle qui présente l’erreur de généralisation la plus faible alors que nous avons accès seulement à l’erreur d’apprentissage ? Regardons d’abord si la réduction de l’erreur d’apprentissage permet de diminuer l’erreur de généralisation.
Pour cela, considérons des données de test, non utilisées pour l’apprentissage mais disposant de l’information de supervision. Comparons trois modèles différents, chacun appartenant à une famille paramétrique distincte :
Un modèle linéaire obtenu par analyse factorielle discriminante (AFD).
Un perceptron multicouches (PMC) avec un coefficient « d’oubli » (weight decay) \(\alpha = 10^{-5}\).
Un perceptron multicouches (PMC) avec un coefficient « d’oubli » \(\alpha = 1\).
Le premier modèle est obtenu en optimisant les paramètres afin de maximiser un critère discriminant spécifique (qui, sous certaines hypothèses, correspond à la minimisation du risque empirique). Les deux derniers modèles sont obtenus en optimisant les paramètres afin de minimiser l’erreur d’apprentissage (le risque empirique).
Les deux figures suivantes montrent l’erreur d’apprentissage pour chacun de ces modèles et respectivement leurs erreurs sur les données de test (vues comme des estimations de leurs erreurs de généralisation).
Fig. 15 Comparaison entre frontières de discrimination et entre erreurs d’apprentissage pour trois modèles¶
Fig. 16 Comparaison entre frontières de discrimination et entre erreurs de test pour trois modèles¶
En comparant les erreurs obtenues par les différents modèles sur les données d’apprentissage et sur les données de test nous pouvons faire plusieurs constats :
Le modèle qui a la plus faible erreur d’apprentissage n’est pas celui qui présente la plus faible erreur de test. Une réduction de l’erreur d’apprentissage n’a pas nécessairement comme conséquence une diminution de l’erreur de généralisation. Cela reste valable en comparant des modèles issus de la même famille paramétrique, par ex. par early stopping (arrêt précoce de la procédure d’optimisation) pour les PMC.
L’erreur d’apprentissage est une estimation optimiste de l’erreur de test. Ce constat peut être généralisé.
L’écart entre l’erreur d’apprentissage et l’erreur de test d’un modèle dépend de la famille paramétrique dont il fait partie. Dans l’exemple considéré, l’écart est le plus élevé pour le modèle le plus « complexe », le PMC avec \(\alpha = 10^{-5}\). Nous verrons dans la suite (section Comment mesurer la capacité ?) que ce constat peut aussi être généralisé.
Il n’est pas possible de mesurer l’erreur de généralisation, pouvons-nous alors au moins l”estimer ? Oui, mais chaque approche présente un inconvénient significatif :
Le risque espéré peut être estimé par l’erreur sur des données de test, pour lesquelles l’information de supervision est disponible mais qui n’ont pas été utilisées pour l’apprentissage. Les observations disponibles avec information de supervision sont donc séparées en données d’apprentissage (permettant d’obtenir le modèle) et données de test (permettant d’estimer sa capacité de généralisation). Cela a pour conséquence une réduction significative du volume de données disponibles pour l’apprentissage.
Sous certaines conditions, il est possible de déterminer une borne supérieure finie sur l’écart entre l’erreur d’apprentissage et l’erreur de généralisation. Nous verrons plus loin (section Comment mesurer la capacité ?) une telle borne qui dépend des caractéristiques de la famille paramétrique considérée. Si l’erreur d’apprentissage est connue (par ex. 4%) et la borne calculée est faible (par ex. 5%), nous obtenons alors une borne supérieure potentiellement intéressante (par ex. 4% + 5% = 9%) sur l’erreur de généralisation du modèle. Malheureusement, rares sont les situations pratiques permettant d’atteindre des bornes suffisamment étroites pour être utiles.
Modélisation à partir de données : un cadre plus précis¶
Après cette première présentation qui met en évidence quelques-une des questions posées par la modélisation décisionnelle à partir de données, nous devons définir un cadre plus précis dans lequel donner des réponses.
Pour commencer, voici les étapes générales de la modélisation décisionnelle à partir de données :
Préparation des données et choix d’une fonction de perte permettant de qualifier les réponses d’un modèle.
Choix des familles paramétriques dans lesquelles les modèles seront recherchés.
Dans chaque famille paramétrique, estimation du « meilleur » modèle intra-famille.
Choix du meilleur modèle entre familles.
Évaluation des performances de généralisation du modèle retenu.
Ces étapes seront étudiées de façon plus détaillée dans la suite.
Le domaine dans lequel sont définies les variables explicatives (ou l’espace d’entrée) est noté \(\mathcal{X}\). Par exemple, pour des observations décrites par \(p\) variables numériques à valeurs réélles, \(\mathcal{X} \subseteq \mathbb{R}^p\).
Le domaine dans lequel est définie la variable expliquée (ou l’espace de sortie) est noté \(\mathcal{Y}\). Par exemple, pour un problème de classement à deux classes nous pouvons avoir \(\mathcal{Y} = \{-1;1\}\) et pour un problème de régression \(\mathcal{Y} \subseteq \mathbb{R}\).
Chaque observation (ou donnée) est définie par des valeurs pour les variables explicatives et une valeur pour la variable expliquée. Pour un problème donné, l’observation générique est décrite par les variables aléatoires \((X,Y) \in \mathcal{X} \times \mathcal{Y}\) suivant la distribution (inconnue a priori) \(P\). Une observation est donc \((\mathbf{x}_i, y_i)_{1 \leq i \leq N}\), \(\mathbf{x}_i \in \mathcal{X}\), \(y_i \in \mathcal{Y}\). L’information de supervision est la valeur de la variable expliquée, \(y_i \in \mathcal{Y}\). Un ensemble de \(N\) observations avec information de supervision est noté \(\mathcal{D}_N = \{(\mathbf{x}_i, y_i)\}_{1 \leq i \leq N}\).
Examinons deux exemples concrets. Le premier montre un problème de classement en deux classes de données bidimensionnelles (voir la figure suivante). Dans ce cas, \(\mathcal{X} \subset \mathbb{R}^2\) et \(\mathcal{Y} = \{c_1, c_2\}\) (\(c_1, c_2\) sont les deux étiquettes de classe).
Fig. 17 Ensemble d’observations bidimensionnelles dans un problème de classement en deux classes : \(\mathcal{X} \subset \mathbb{R}^2\), \(\mathcal{Y} = \{c_1, c_2\}\) (\(c_1\) représentée par la couleur verte, \(c_1\) par la couleur rouge)¶
Le second exemple montre un problème de régression entre variables unidimensionnelles (voir la figure suivante). Dans ce cas, \(\mathcal{X} \subset \mathbb{R}\), \(\mathcal{Y} \subset \mathbb{R}\).
Fig. 18 Ensemble d’observations dans un problème de régression entre deux variables unidimensionnelles : \(\mathcal{X} \subset \mathbb{R}\), \(\mathcal{Y} \subset \mathbb{R}\).¶
Sauf cas particuliers (par ex. des données décrivant des séries temporelles), on considère les observations de \(\mathcal{D}_N\) issues de tirages indépendants (le tirage \(i\) est indépendant du tirage \(j\) pour \(i \neq j\)) et identiquement distribués suivant la distribution (inconnue a priori) \(P\).
L’objectif de la modélisation décisionnelle est de trouver une fonction (un modèle) \(f \in \mathcal{F}\), \(f : \mathcal{X} \rightarrow \mathcal{Y}\), qui prédit \(y \in \mathcal{Y}\) à partir de \(\mathbf{x} \in \mathcal{X}\) et présente le risque espéré \(R(f) = E_P[L(X,Y,f)]\) le plus faible (c’est à dire, l’erreur de généralisation la plus faible). Par \(L()\) nous avons noté la fonction de perte (ou d’erreur) qui qualifie dans quelle mesure \(f(\mathbf{x})\) correspond à la valeur attendue de \(y\). \(E_P\) est ici l”espérance par rapport à la distribution inconnue \(P\).
Le choix d’une fonction de perte dépend de la nature du problème de modélisation (classement, régression ou prédiction structurée), du choix de la famille de fonctions \(\mathcal{F}\) dans laquelle le modèle \(f\) est recherché, ainsi que de la procédure d’optimisation permettant de trouver \(f\) dans \(\mathcal{F}\). Nous examinons dans la suite les fonctions de perte les plus utilisées, pour des problèmes de classement ou de régression.
Pour des problèmes de classement (ou de discrimination entre classes), la fonction de perte la plus fréquemment rencontrée est la perte 0-1 (0-1 loss), définie de la façon suivante :
Si les valeurs \(f(\mathbf{x})\) font partie (comme \(y\)) du domaine de valeurs de la variable expliquée, \(f(\mathbf{x}), y \in \mathcal{Y}\), domaine qui doit être un ensemble fini (par ex. \(\mathcal{Y} = \{-1;1\}\)), alors \(L_{01}(\mathbf{x},y,f) = \mathbf{1}_{f(\mathbf{x}) \neq y}\), où \(\mathbf{1}_{f(\mathbf{x}) \neq y} = \left\{\begin{array}{ll}1&\textrm{si} \, f(\mathbf{x}) \neq y\\0&\textrm{sinon}\end{array}\right.\).
Si \(f(\mathbf{x}) \in \mathbb{R}\) et \(\mathcal{Y} = \{-1;1\}\) (classement en deux classes) alors \(L_{01}(\mathbf{x},y,f) = \mathbf{1}_{H(f(\mathbf{x})) \neq y}\), où \(H(z) = \left\{\begin{array}{ll}+1&\textrm{si} \, z \geq 0\\-1&\textrm{sinon}\end{array}\right.\) est la fonction échelon adéquate.
Fig. 19 Exemple de classement avec une frontière linéaire. Les flèches bleues indiquent quelques données mal classées par le modèle¶
La perte (l’erreur) est donc nulle si la prédiction est correcte (ou a le bon signe) et égale à 1 si la prédiction est incorrecte (ou n’a pas le bon signe). Cette définition a plusieurs implications, notamment :
Toute erreur de classement apporte une même pénalité (égale à 1), quelle que soit cette erreur. Par exemple, le classement erroné d’un patient sain dans la classe des malades apporte une même pénalité que le classement erroné d’un malade dans la classe des patients sains. Nous pouvons constater que cette perte (erreur, cout) symétrique n’est pas toujours un choix satisfaisant. Des pertes asymétriques peuvent être facilement définies.
Pour \(f(\mathbf{x}) \in \mathbb{R}\), seul compte le signe de \(f(\mathbf{x})\). Deux prédictions \(f(\mathbf{x}) = 0,001\) et \(f(\mathbf{x})=1000\) sont parfaitement équivalentes avec cette fonction de perte, or ces deux prédictions n’ont peut-être pas une même signification pour le problème modélisé.
Une autre fonction de perte employée fréquemment dans des problèmes de classement en deux classes avec maximisation de la marge (voir le premier cours sur les SVM) est la perte hinge loss, définie pour \(f(\mathbf{x}) \in \mathbb{R}\) de la façon suivante : \(L_h(\mathbf{x},y,f) = \max\{0, 1 - y f(\mathbf{x})\}\), voir aussi la figure suivante :
Fig. 20 Hinge loss pour \(y = -1\) (en rouge) et \(y = 1\) (en bleu)¶
Nous pouvons remarquer que \(L_h\) n’est pas différentiable par rapport à \(f\) mais admet un sous-gradient, ce qui a un impact sur l’algorithme d’optimisation des paramètres du modèle. Enfin, des extensions de hinge loss ont été définies pour le classement multi-classe et la prédiction structurée, ces extensions seront étudiées ultérieurement.
Pour des problèmes de régression (), la fonction de perte la plus fréquemment rencontrée est la perte (ou l’erreur) quadratique, définie par : \(L_q(\mathbf{x},y,f) = [f(\mathbf{x})-y]^2\) , où \(f(\mathbf{x})\) est la prédiction du modèle \(f\) pour l’entrée \(\mathbf{x}\) et \(y\) est l’information de supervision (la prédiction désirée) pour l’entrée \(\mathbf{x}\). Cette fonction est différentiable par rapport à \(f(\mathbf{x})\) ; si \(f\) est à son tour différentiable par rapport aux paramètres du modèle, alors une optimisation basée sur le gradient peut être appliquée directement, comme nous le verrons plus loin.
Fig. 21 Exemple de régression linéaire entre variables unidimensionnelles. Les traits rouges représentent des écarts entre trois prédictions d’un modèle (linéaire, dans ce cas) et les prédictions désirées correspondantes¶
La seconde étape dans la démarche de modélisation décisionnelle à partir de données est le choix des familles paramétriques dans lesquelles les modèles seront recherchés. Ces familles paramétriques peuvent être :
Les modèles linéaires, pour lesquels la prédiction est obtenue à partir d’une combinaison linéaire des variables explicatives.
Par exemple, pour un problème de classement, la prédiction pour l’entrée \(\mathbf{x}\) est le résultat de l’application de la fonction échelon \(H() \in \{-1, 1\}\) à la sortie du modèle \(f(\mathbf{x}) = \mathbf{w}^T \mathbf{x} + w_0\), qui est une combinaison linéaire des composantes du vecteur \(\mathbf{x}\) avec les pondérations du vecteur \(\mathbf{w}\) et le terme libre \(w_0\).
Fig. 22 Modèle linéaire pour le classement à deux classes d’observations bidimensionnelles¶
Pour un problème de régression entre variables unidimensionnelles, la prédiction pour l’entrée \(\mathbf{x}\) est \(f(x) = w_1 x + w_0\) et présente donc uné dépendance linéaire de \(\mathbf{x}\).
Fig. 23 Modèle linéaire pour la régression entre variables unidimensionnelles¶
Les modèles linéaires peuvent s’avérer insuffisants dans l’explication de la variable de sortie (ou variable « à expliquer ») à partir d’une combinaison linéaire des variables explicatives. Leur capacité d’approximation d’une frontière de discrimination dans le cas d’un problème de classement ou d’une dépendance dans le cas d’un problème de régression peut être trop limitée. Dans l’exemple de problème de classement ci-dessus (Fig. 22), les classes ne sont pas linéairement séparables : la meilleure séparation linéaire présente une erreur empirique relativement élevée (ici d’env. 12%). Commencer par les modèles linéaires est une bonne pratique.
Les modèles polynomiaux de degré \(n\) borné (\(n > 1\) pour aller au-delà des modèles linéaires). Dans ce cas, la dépendance entre l’entrée (les variables explicatives) et la prédiction fournie par le modèle est polynomiale. Chaque valeur de borne sur le degré \(n\) définit une famille paramétrique. La capacité d’approximation des polynomes augmente avec leur degré.
Diverses familles de modèles non linéaires, par exemple les perceptrons multicouches (PMC) d’architecture donnée, etc.
Vu que les familles les plus « simples » (par ex. les modèles linéaires) peuvent s’avérer insuffisantes, on peut se demander s’il faut encore les considérer ou alors s’intéresser directement (et systématiquement) à une famille dont la capacité d’approximation est aussi grande que possible. Revenons à l’exemple antérieur (voir Fig. 15, Fig. 16) d’un problème de classement traité avec trois familles de modèles : modèles linéaires, PMC avec \(\alpha = 10^{-5}\) et PMC avec \(\alpha = 1\). De ces trois familles, les modèles linéaires présentent la capacité d’approximation la plus faible et les PMC avec \(\alpha = 10^{-5}\) la capacité d’approximation la plus élevée. La figure suivante reprend les solutions trouvées dans chaque famille, avec les erreurs d’apprentissage et de généralisation correspondantes (l’erreur de généralisation est estimée par l’erreur sur les données de test).
Fig. 24 Capacité d’approximation, erreur d’apprentissage et erreur de généralisation pour trois modèles issus de la famille linéaire (à gauche), PMC avec \(\alpha = 10^{-5}\) (centre) et PMC avec \(\alpha = 1\) (droite)¶
Il est facile de constater que, si l’erreur d’apprentissage obtenue est la plus faible pour le modèle issu de la famille avec la capacité d’approximation la plus élevée (PMC avec \(\alpha = 10^{-5}\)), l’erreur de généralisation est la plus faible pour un modèle issu d’une autre famille (PMC avec \(\alpha = 1\)). Une capacité d’approximation trop élevée peut mener au sur-apprentissage (overfitting) ou apprentissage « par cœur » : l’erreur d’apprentissage est très faible mais l’erreur sur les données de test est comparativement élevée ; le modèle a appris les particularités (par ex. le bruit) des données d’apprentissage.
Ce n’est donc pas nécessairement dans la famille qui a la capacité la plus grande qu’on trouve le modèle qui généralise le mieux. Examiner des familles de capacité inférieure n’est pas sans intérêt. Le lien entre la capacité de la famille de modèles et la généralisation obtenue sera exploré dans la suite.
Il est important maintenant de s’intéresser de plus près à la façon dont un modèle est estimé au sein d’une famille paramétrique considérée.
Rappelons que notre objectif est de trouver, dans une famille \(\mathcal{F}\) choisie, une fonction (un modèle) \(f : \mathcal{X} \rightarrow \mathcal{Y}\) qui prédit \(y\) à partir de \(\mathbf{x}\) et présente le risque espéré (ou théorique) \(R(f) = E_P[L(X,Y,f)]\) le plus faible. Or, ce risque espéré \(R(f)\) ne peut pas être évalué car \(P\) est inconnue. Mais nous pouvons mesurer le risque empirique \(R_{\mathcal{D}_N}(f) = \frac{1}{N} \sum_{i=1}^N{L(\mathbf{x}_i,y_i,f)}\) sur les données d’apprentissage \(\mathcal{D}_N\) (l’erreur d’apprentissage). La recherche du meilleur modèle au sein de la famille \(\mathcal{F}\), ne peut tenir compte directement que de ce risque empirique. Nous pouvons mentionner trois approches qui seront détaillées dans des sous-sections ultérieures :
La minimisation du risque empirique (MRE) : recherche du modèle qui minimise l’erreur d’apprentissage, \(f^*_{\mathcal{D}_N} = \arg \min_{f \in \mathcal{F}} R_{\mathcal{D}_N}(f)\).
La minimisation du risque empirique régularisé (MRER) : recherche du modèle qui minimise la somme entre l’erreur d’apprentissage et un terme de régularisation\(G(f)\) pondéré par une constante (\(\alpha\)), \(f^*_{\mathcal{D}_N} = \arg \min_{f \in \mathcal{F}} [R_{\mathcal{D}_N}(f) + \alpha G(f)]\).
La minimisation du risque structurel (MRS) : on considère une séquence de familles de capacité qui augmente et on effectue une estimation MRE dans chaque famille. Le choix final d’un modèle tient compte à la fois du risque empirique \(R_{\mathcal{D}_N}\) du modèle et de la capacité d’approximation de la famille dont il est issu.
La compréhension du lien entre la capacité d’approximation d’une famille de modèles et les perfomances de généralisation qu’elle permet d’atteindre est facilité par une analyse des composantes du risque espéré. Considérons les notations suivantes :
\(f^*_{\mathcal{D}_N}\) est la fonction de \(\mathcal{F}\) qui minimise le risque empirique \(R_{\mathcal{D}_N}\),
\(f^*\) est la fonction de \(\mathcal{F}\) qui minimise le risque espéré \(R\).
Nous pouvons alors écrire (addition et soustraction de \(R^*\) et respectivement de \(R(f^*)\), ensuite regroupement) :
Dans cette égalité, \(R^*\) est le risque résiduel (ou risque de Bayes), une borne inférieure pour le risque espéré de tout modèle, quelle que soit la famille dont il est issu. Ce risque résiduel est strictement non nul en présence de bruit, car à une observation \(\mathbf{x}\) dont les composantes (les valeurs des variables explicatives) sont entachées de bruit (ou d’incertitude de mesure) peuvent correspondre différentes valeurs de \(y\). Un risque espéré égal au risque résiduel ne peut être atteint que par le meilleur modèle toutes familles confondues.
La seconde composante du membre droit, \([R(f^*) - R^*]\), est l’erreur d’approximation. Elle est égale à zéro pour la (ou les) famille(s) qui contien(nen)t le meilleur modèle toutes familles confondues. Pour les familles qui ne contiennent pas ce modèle, l’erreur d’approximation est strictement positive. Si une famille \(\mathcal{F}_1\) a une capacité supérieure à une autre famille \(\mathcal{F}_2\), le meilleur modèle de \(\mathcal{F}_1\) (noté \(f^*_1\)) présente une meilleure (= plus faible) erreur d’approximation que le meilleur modèle de \(\mathcal{F}_2\) (noté \(f^*_2\)).
La dernière composante du membre droit, \([R(f^*_{\mathcal{D}_N}) - R(f^*)]\), est l’erreur d’estimation qui met en évidence le fait que la recherche du meilleur modèle dans une famille donnée ne peut se faire en tenant compte directement de la minimisation du risque espéré. Elle est égale à zéro si la fonction de \(\mathcal{F}\) qui minimise le risque empirique est également celle qui minimise le risque espéré. Plus la capacité d’une famille \(\mathcal{F}\) est élevée, plus il est probable que le modèle qui minimise le risque empirique (sur des données d’apprentissage qui correspondent à un échantillon de taille \(N\)) s’éloigne du modèle qui minimise le risque espéré ; ainsi, l’erreur d’estimation augmente.
Pour des familles de modèles de capacité croissante, l’erreur d’approximation diminue (il devient plus probable de trouver le meilleur modèle toutes familles confondues dans la famille considérée) mais l’erreur d’estimation augmente. La borne inférieure restant la même (le risque résiduel), la meilleure famille est celle pour laquelle la somme entre l’erreur d’aproximation et l’erreur d’estimation est la plus faible.
Il est important de remarquer qu’une analyse similaire des composantes du risque espéré peut être obtenue en remplaçant \(f^*_{\mathcal{D}_N}\) par une fonction trouvée par une autre procédure que la minimisation du risque empirique (MRE).
Examiner un exemple est utile pour mieux apprécier le lien entre la capacité d’approximation, l’erreur d’approximation et l’erreur d’estimation. Considérons le même problème et les mêmes trois familles de modèles que pour la Fig. 24. La figure suivante montre les modèles obtenus au sein de chaque famille pour trois échantillons d’apprentissage différents (tous de même nombre d’observations), les moyennes de leurs erreurs d’apprentissage, ainsi que les moyennes et écart-types de leurs erreurs de test.
Fig. 25 Liens entre capacité, erreur d’approximation et erreur d’estimation¶
La capacité d’aproximation de la famille linéaire (à gauche) est inférieure à celle de la famille PMC avec \(\alpha = 1\) (à droite) qui est, à son tour, inférieure à la capacité de la famille PMC avec \(\alpha = 10^{-5}\) (au centre). Nous pouvons faire les observations suivantes :
Pour la famille linéaire (les modèles sont obtenus ici par AFD) la moyenne des erreurs d’apprentissage est élevée, la capacité peut être jugée insuffisante pour ce problème où les classes ne sont pas linéairement séparables. Les modèles linéaires ne peuvent pas approcher suffisamment le meilleur modèle toutes familles confondues, l’erreur d’approximation est élevée (il y a un fort biais).
Pour la famille définie par les PMC avec \(\alpha = 10^{-5}\), la moyenne des erreurs d’apprentissage est très faible, donc l’erreur d’approximation est potentiellement faible, la capacité peut être jugée suffisante. En revanche, la moyenne des erreurs de test est bien plus élevée que la moyenne des erreurs d’apprentissage et la variance des erreurs de test est élevée (supérieure à celle de l’autre famille de PMC, avec \(\alpha = 1\)), ce qui indique que l’erreur d’estimation est élevée.
Pour la famille définie par les PMC avec \(\alpha = 1\), la moyenne des erreurs de test est comparativement faible, ce qui indique une somme assez faible entre erreur d’approximation et erreur d’estimation. La généralisation est meilleure que celle obtenue avec les deux autres familles. Enfin, la moyenne des erreurs de test est assez faible et proche de la moyenne des erreurs d’apprentissage.
Jusqu’ici nous avons parlé de la capacité (d’approximation) d’une famille de modèles de façon intuitive, sans définir une mesure précise. Nous présentons brièvement dans cette section la mesure de capacité la plus connue, la dimension de Vapnik-Chervonenkis, qui permet d’obtenir plusieurs résultats intéressants (voir aussi le chapitre sur les SVM).
Pour introduire cette mesure, considérons un ensemble de \(N\) vecteurs \(\{\mathbf{x}_i\}_{1 \leq i \leq N} \in \mathbb{R}^p\) (vecteurs à composantes réelles, de dimension \(p\)). Il y a \(2^N\) façons différentes de séparer cet ensemble en deux parties (attention, l’ensemble vide et l’ensemble complet sont aussi des parties). Nous pouvons maintenant évaluer la capacité d’une famille \(\mathcal{F}\) de fonctions \(f : \mathbb{R}^p \rightarrow \{-1, 1\}\) en examinant ses capacités à séparer différents ensembles de vecteurs. Une séparation est « matérialisée » par le fait qu’une fonction \(f \in \mathcal{F}\) associe la valeur \(-1\) à certains vecteurs et la valeur \(1\) aux autres.
Définition : la famille \(\mathcal{F}\) de fonctions \(f : \mathbb{R}^p \rightarrow \{-1, 1\}\)pulvérise\(\{\mathbf{x}_i\}_{1 \leq i \leq N}\) si toutes les \(2^N\) séparations peuvent être construites avec des fonctions de \(\mathcal{F}\).
Définition : l’ensemble \(\mathcal{F}\) est de VC-dimension (dimension de Vapnik-Chervonenkis) \(h\) s’il pulvérise au moins un ensemble de \(h\) vecteurs et aucun ensemble de \(h+1\) vecteurs.
Par exemple, la VC-dimension de l’ensemble des hyperplans (les séparations linéaires de dimension \(p-1\)) de \(\mathbb{R}^p\) est \(h = p+1\). Pour illustrer, considérons le cas particulier de l’espace bidimensionnel \(\mathbb{R}^2\). Dans \(\mathbb{R}^2\) les hyperplans sont les droites. Dans la figure suivante nous pouvons voir un triplet et un quadruplet de points (vecteurs de \(\mathbb{R}^2\)).
Fig. 26 Triplet et quadruplet de points dans \(\mathbb{R}^2\)¶
L’ensemble des droites de \(\mathbb{R}^2\) pulvérise le triplet de points à gauche car pour chacune des \(2^3 = 8\) séparations envisageables (\(-1\) associé à tous les points et \(1\) à aucun, \(1\) associé à tous les points et \(-1\) à aucun, \(-1\) associé au point de gauche et \(1\) aux autres, etc.) il existe au moins une droite qui permet de l’obtenir. En revanche, on peut montrer qu’aucun quadruplet de points ne peut être pulvérisé (il existe au moins une séparation qui ne peut pas être faite avec une droite). Par exemple, pour le quadruplet représenté dans la figure, aucune droite ne peut séparer les points bleus des points rouges.
La VC-dimension est une mesure intéressante de la capacité car elle permet, entre autres, d’obtenir une borne pour l’écart entre le risque théorique (ou risque espéré) et le risque empirique.
Théorème[BBL04] : soit \(R_{\mathcal{D}_N}(f)\) le risque empirique défini par la fonction de perte \(L_{01}(\mathbf{x},y,f) = \mathbf{1}_{f(\mathbf{x}) \neq y}\); si la VC-dimension de \(\mathcal{F}\) est \(h < \infty\) alors pour toute \(f \in \mathcal{F}\), avec une probabilité au mois égale à \(1-\delta\) (\(0 < \delta < 1\)), on a
\[R(f) \leq R_{\mathcal{D}_N}(f) + \underbrace{\sqrt{\frac{h \left(\log\frac{2N}{h} + 1\right) -\log\frac{\delta}{4}}{N}}}_{B(N,\mathcal{F})} \quad \textrm{pour} \quad N > h
\]
La forme de \(B(N,\mathcal{F})\) permet de constater que la borne diminue quand le nombre de données d’apprentissage \(N\) augmente. En effet, avec plus de données d’apprentissage il est possible de mieux « contraindre » le modèle appris et de réduire ainsi la composante « estimation » de l’erreur.
La borne a également une valeur plus faible pour une famille \(\mathcal{F}\) de VC-dimension \(h\) plus faible. Avec un nombre fixé de données d’apprentissage il est possible de mieux « contraindre » un modèle moins « flexible ». Malheureusement, une famille avec la VC-dimension \(h\) trop faible ne sera pas capable de faire la séparation attendue, la composante « approximation » de l’erreur sera trop élevée.
Enfin, la borne peut être réduite par l’augmentation de \(\delta\) (l’écart par rapport à une probabilité de 1), c’est à dire par une diminution des « garanties » qu’apporte la borne.
Il est important de remarquer que cette borne \(B(N,\mathcal{F})\) ne fait pas intervenir le nombre de variables (la dimension de l’espace dans lequel se trouvent les observations), ni la loi conjointe (inconnue) \(P\). En ce sens il s’agit d’un résultat dans le pire des cas, intéressant d’un point de vue théorique mais malheureusement peu utile en pratique car pour obtenir une borne utile (de valeur faible) il faudrait en général disposer d’un nombre \(N\) très élevé d’observations pour l’apprentissage.
Nous pouvons faire une synthèse des conséquences de l’existence d’une borne, \(R(f) \leq R_{\mathcal{D}_N}(f) + B(N,\mathcal{F})\), et de la forme de cette borne \(B(N,\mathcal{F})\) pour des familles de modèles de capacité différente :
Pour une famille \(\mathcal{F}\) de capacité trop faible (par ex. ici les modèles linéaires) la borne \(B(N,\mathcal{F})\) est basse mais comme le risque empirique (l’erreur d’apprentissage) \(R_{\mathcal{D}_N}(f)\) est élevé(e) nous ne pouvons pas obtenir de garantie intéressante pour le risque espéré \(R(f)\) (pour les performances de généralisation).
Pour une famille \(\mathcal{F}\) de capacité trop élevée (par ex. ici PMC avec \(\alpha = 10^-5\)) le risque empirique \(R_{\mathcal{D}_N}(f)\) est vraisemblablement faible (cette famille a une capacité d’approximation élevée) mais la valeur de la borne \(B(N,\mathcal{F})\) est également élevée. Dans ce cas encore nous ne pouvons pas obtenir de garantie intéressante pour le risque espéré \(R(f)\).
Pour une famille \(\mathcal{F}\) de capacité « adéquate » (par ex. ici PMC avec \(\alpha = 1\)) le risque empirique \(R_{\mathcal{D}_N}(f)\) reste faible et la valeur de la borne \(B(N,\mathcal{F})\) est plutôt basse, l’inégalité \(R(f) \leq R_{\mathcal{D}_N}(f) + B(N,\mathcal{F})\) peut donc nous donner une garantie intéressante pour le risque espéré \(R(f)\) ! Rappelons ici qu’une borne comme celle obtenue dans le théorème précédent est toutefois rarement utile en pratique car elle peut être basse seulement si on dispose de très nombreuses données d’apprentissage (\(N\) très élevé).
Minimisation du risque empirique régularisé (MRER)¶
Les sections précédentes nous ont permis de constater que la minimisation du risque empirique (MRE) n’était pas suffisante pour assurer une bonne généralisation pour le modèle obtenu. Si la famille de modèles possède une capacité trop élevée, la minimisation du risque empirique a pour conséquence un « apprentissage par cœur » (overfitting). Afin d’éviter ce problème il est nécessaire de maîtriser la capacité de \(\mathcal{F}\) (ou la complexité du modèle).
Une approche de maîtrise implicite de la capacité est la régularisation : le modèle \(f^*_{\mathcal{D}_N}\) optimal au sein de la famille \(\mathcal{F}\) considérée est obtenu en minimisant non simplement le risque empirique, mais plutôt la somme entre le risque empirique \(R_{\mathcal{D}_N}(f)\) et un terme \(G(f)\) qui pénalise (indirectement) la capacité
Ici \(\alpha\) est un hyper-paramètre qui pondère le terme de régularisation. Plus la valeur de \(\alpha\) est élevée, plus la pénalité de la capacité est importante par rapport à l’erreur empirique.
Le terme de régularisation \(G(f)\) peut prendre différentes formes, en rapport aussi avec le choix de la famille \(\mathcal{F}\) dans laquelle le modèle est recherché. En règle générale, \(G(f)\) doit augmenter avec la « capacité » de \(\mathcal{F}\), mais \(G(f)\) ne fait que rarement explicitement référence une mesure de capacité formellement définie. Voici deux exemples de méthodes de régularisation couramment employées avec les réseaux de neurones :
« Oubli » (weight decay) : \(G(f) = \|\mathbf{w}\|_2^2\), \(\mathbf{w}\) étant le vecteur de paramètres du modèle (réseau de neurones, par ex. perceptron multi-couches).
Régularisation par arrêt précoce (early stopping) : aucun terme \(G(f)\) n’est présent mais l’algorithme d’optimisation (non linéaire, pour les perceptrons multi-couches) s’arrête avant d’atteindre le minimum du risque empirique.
La capacité de la famille de modèles peut également être maîtrisée de façon explicite dans le cadre de la minimisation du risque structurel (voir par ex. [BBL04]). La modélisation décisionnelle passe dans ce cas par les étapes suivantes :
Définition d’une séquence \(\mathcal{F}_1 \subset \mathcal{F}_2 \subset \mathcal{F}_3 \ldots\) de familles de capacités croissantes, c’est à dire pour lesquelles \(h_1 < h_2 < h_3 \ldots\).
Minimisation dans chaque famille du risque empirique \(f^{(i)*}_{\mathcal{D}_N} = \arg \min_{f \in \mathcal{F}_i} R_{\mathcal{D}_N}(f)\), pour \(i \in \{1, 2, 3 \ldots\}\).
En tenant compte de la borne trouvée pour le risque espéré \(R\), sélection de \(f^{(i)*}_{\mathcal{D}_N}, i \in \{1, 2, 3 \ldots\}\), qui permet de minimiser la somme entre le risque empirique et la borne de généralisation correspondant à cette famille : \(R_{\mathcal{D}_N}(f^{(i)*}_{\mathcal{D}_N}) + B(N,\mathcal{F}_i)\).
Comment minimiser le risque empirique (régularisé) ?¶
A l’intérieur d’une famille paramétrique \(\mathcal{F}\), un modèle est défini par les valeurs d’un ensemble de paramètres, par exemple :
Pour un modèle linéaire pour la régression entre deux variables unidimensionnelles, \(y = a x + b\), les paramètres sont \(a\) (le pente de la droite qui représente le modèle, voir la figure suivante) et \(b\) (l’ordonnée du point où la droite intersecte l’axe vertical, ou intercept).
Pour un perceptron multi-couches d’architecture donnée, les paramètres sont les poids des connexions et les seuils des neurones de la (des) couche(s) cachée(s) et de la couche de sortie.
Le modèle recherché \(f^*_{\mathcal{D}_N}\) au sein de la famille paramétrique \(\mathcal{F}\) est défini par les valeurs des paramètres qui optimisent le critère retenu (par ex. MRER). Afin de trouver ces valeurs optimales, suivant la famille paramétrique et le critère optimisé, nous pouvons rencontrer :
Une solution analytique directe, permettant d’obtenir les valeurs recherchées comme solution d’un système d’équations. C’est un cas assez rare, rencontré notamment pour certains modèles linéaires.
Des algorithmes itératifs, par exemple
Optimisation quadratique sous contraintes d’inégalités, employée notamment pour les machines à vecteurs de support (SVM). Sous certaines conditions, l’atteinte de l’optimum global est garantie.
Optimisation non linéaire plus générale, employée notamment pour les réseaux de neurones à une couche cachée ou plus profonds. En général la convergence vers un optimum global ne peut être garantie, la solution atteinte sera donc sous-optimale.
Nous examinerons de plus près ces méthodes d’optimisation ultérieurement, lors de l’étude de différentes familles de modèles. Il est toutefois utile de voir déjà un exemple simple d’obtention d’une solution analytique et nous considérons pour cela le cas de la régression linéaire.
Pour le problème de régression, chaque observation est caractérisée par les valeurs de \(p\) variables explicatives réelles et la valeur d’une variable expliquée réelle. L’espace d’entrée est \(\mathcal{X} = \mathbb{R}^p\) et l’espace de sortie \(\mathcal{Y} = \mathbb{R}\). Pour trouver le modèle nous disposons de \(N\) observations qui possèdent l’information de supervision (les valeurs de la variable expliquée), \(\mathcal{D}_N = \{(\mathbf{x}_i, y_i)\}_{1 \leq i \leq N}\).
Nous cherchons le modèle dans la famille de modèles linéaires \(\hat{y} = w_0 + \sum_{j=1}^p w_j x_{ji}\), où par \(\hat{y}\) nous avons noté la prédiction du modèle pour l’entrée \(\mathbf{x}\). Un modèle est défini par les valeurs des \(p+1\) paramètres \(w_j\), \(0 \leq j \leq p\).
Sous forme matricielle, la prédiction du modèle est \(\hat{\mathbf{y}} = \mathbf{X} \mathbf{w}\), où \(\mathbf{X}\) est la matrice \(N \times (p+1)\) dont les lignes sont les observations de \(\mathcal{D}_N\) et les colonnes correspondent aux variables, sauf pour la dernière qui est une colonne de \(1\) et permet d’inclure \(w_0\) dans \(\mathbf{w}\).
La recherche du modèle linéaire (défini par le vecteur de paramètres \(\mathbf{w}^*\)) peut suivre une de ces deux approches :
Approche de minimisation du risque empirique (MRE) : l’erreur quadratique totale \(\sum_{i=1}^N (\hat{y}_i - y_i)^2\) sur \(\mathcal{D}_N\).
La solution est obtenue par calcul direct : \(\mathbf{w}^* = \mathbf{X}^+ \mathbf{y}\), où \(\mathbf{X}^+\) est la pseudo-inverse Moore-Penrose de \(\mathbf{X}\).
Si \(\mathbf{X}^T \mathbf{X}\) est inversible, alors \(\mathbf{X}^+ = (\mathbf{X}^T \mathbf{X})^{-1} \mathbf{X}^T\).
Approche de minimisation du risque empirique régularisé (MRER) : la somme entre l’erreur quadratique sur \(\mathcal{D}_N\) et un terme de régularisation. Par exemple, dans le cas particulier de la régularisation de Tikhonov, \(\sum_{i=1}^N (\hat{y}_i - y_i)^2 + \|\mathbf{w}\|^2_2\).
La solution est, dans ce cas, \(\mathbf{w}^* = (\mathbf{X}^T \mathbf{X} + \mathbf{I}_{p+1})^{-1} \mathbf{X}^T \mathbf{y}\), où \(\mathbf{I}_{p+1}\) est la matrice unité de rang \(p+1\).
Modélisation décisionnelle : que faut-il retenir ?¶
Pour construire un modèle décisionnel à partir de données, l’information de supervision est indispensable.
L’ojectif est d’obtenir le modèle qui présente la meilleure généralisation (et non la plus faible erreur d’apprentissage).
L’erreur d’apprentissage (le risque empirique) est excessivement optimiste comme estimateur de l’erreur de généralisation (risque espéré).
Pour minimiser le risque espéré, il faut chercher le bon compromis entre la minimisation de la capacité de la famille de modèles et la minimisation de l’erreur d’apprentissage.
L’estimation directe du risque espéré par le risque empirique (appelée aussi estimation in-sample) est donc excessivement optimiste, surtout pour des familles \(\mathcal{F}\) de capacité élevée (ou infinie), et doit donc être évitée. Lorsque la capacité de \(\mathcal{F}\) est faible et le nombre \(N\) de données d’apprentissage très élevé (situation très rare en pratique), des bornes de généralisation \(B(N,\mathcal{F})\) suffisamment réduites pour majorer de façon utile le risque espéré peuvent éventuellement être obtenues et nous pouvons nous servir de \(R(f^*_{\mathcal{D}_N}) \leq R_{\mathcal{D}_N}(f^*_{\mathcal{D}_N}) + B(N,\mathcal{F})\).
Une méthode générale pour estimer le risque espéré est celle des données de test ou de l”échantillon-test (appelée aussi estimation out-of-sample) :
L’ensemble de données disponibles \(\mathcal{D}_N\) est partitionné en deux ensembles mutuellement exclusifs par sélection aléatoire, les données d’apprentissage \(\mathcal{A}\) (par ex. env. 70% du nombre total) et les données de validation \(\mathcal{V}\) (par ex. 30% du nombre total).
L’apprentissage du modèle est réalisé sur les données de l’ensemble \(\mathcal{A}\), en utilisant une des approches mentionnées (la MRE ou la MRER).
Le risque espéré du modèle résultant est estimé sur les données de \(\mathcal{V}\).
Apprendre sur une partie seulement des données disponibles (\(\mathcal{A}\)) peut être un inconvénient sérieux lorsque \(N\) est faible. Certaines classes peuvent être relativement rares dans les données, diminuer encore le nombre d’observations appartenant à ces classes a un impact négatif sur la qualité du modèle résultant.
Aussi, l’estimateur ainsi obtenu pour le risque espéré a une variance élevée : un autre découpage de \(\mathcal{D}_N\) en \(\mathcal{A}'\) et \(\mathcal{V}'\) peut produire un résultat assez différent. Afin de réduire la variance de l’estimateur il faudrait moyenner les résultats issus de plusieurs découpages différents de \(\mathcal{D}_N\), c’est ce que proposent les méthodes de validation croisée.
Avant d’examiner de plus près ces méthodes, il est important de rappeler qu’un problème de validité important rencontré dans la pratique est la non stationnarité des phénomènes modélisés : les données d’apprentissage deviennent de moins en moins représentatives au fil du temps car la loi conjointe inconnue \(P\) évolue. Il est donc important d’estimer régulièrement l’erreur sur des données récentes afin de mettre en évidence une éventuelle divergence du modèle et donc la nécessité de le mettre à jour.
Pour réduire la variance de l’estimation du risque espéré obtenue sur un échantillon-test, plusieurs partitionnements différents de \(\mathcal{D}_N\) en \(\mathcal{A}_i\) et \(\mathcal{V}_i\) sont réalisés, avec \(i \in {1,\ldots,k}\), \(\mathcal{D}_N = \mathcal{A}_i \cup \mathcal{V}_i\) et \(\mathcal{A}_i \cap \mathcal{V}_i = \varnothing\), à chaque fois un modèle \(f_i\) est appris sur \(\mathcal{A}_i\), son erreur \(L(\mathcal{V}_i,f_i)\) est calculée sur \(\mathcal{V}_i\), et enfin le risque espéré est estimé par la moyenne \(\frac{1}{k} \sum_{i=1}^k L(\mathcal{V}_i,f_i)\). Nous obtenins ainsi un estimateur de variance plus faible, tout en utlisant mieux les données disponibles.
Suivant les partitionnements réalisés, les méthodes peuvent être
Exhaustives, lorsque tous les partitionnements possibles respectant certains effectifs sont utilisés :
Leave\(p\)out (LPO) : \(N-p\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et \(p\) pour la validation (\(\mathcal{V}_i\)), tous les \(C_N^p\) partitionnements possibles avec ces effectifs sont utilisés. Il est donc nécessaire d’apprendre \(C_N^p\) modèles différents, ce qui implique en général un coût excessif.
Leave one out (LOO) : \(N-1\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et une seule pour la validation (\(\|\mathcal{V}_i\| = 1\)), il y a donc \(C_N^1 = N\) partitionnements possibles et il faut apprendre \(N\) modèles différents. Le coût reste élevé pour des données volumineuses (\(N\) élevé).
Non exhaustives :
k-fold : l’ensemble \(\mathcal{D}_N\) est partitionné en \(k\) parties, chaque modèle est évalué sur une des partitions et les \(k-1\) autres sont employées pour son apprentissage. Sont appris \(k\) modèles, chacun évalué sur une partition différente parmi les \(k\). Souvent \(k = 5\) ou \(k = 10\), mais le choix de \(k\) dépend des ressources disponibles car le coût augmente linéairement avec \(k\). La méthode leave one out peut être vue aussi comme un cas particulier de k-fold, pour \(k = N\).
Échantillonnage répété (shuffle and split) : un échantillon aléatoire de \(n\) données est utilisé pour la validation, les autres \(N-n\) données étant employées pour l’apprentissage du modèle, on répète cela \(k\) fois pour obtenir \(k\) modèles. Cette méthode permet d’éviter la contrainte \(k \cdot n = N\), mais présente le désavantage de trouver certaines données dans plusieurs ensembles de validation différents alors que d’autres ne seront présentes dans aucun échantillon.
Même les méthodes non exhaustives sont coûteuses, car il est nécessaire d’apprendre (et ensuite évaluer) \(k\) modèles plutôt qu’un seul. Il faut toutefois noter que ces \(k\) apprentissages (et les \(k\) évaluations correspondantes) peuvent être réalisées en parallèle si nous disposons d’une plate-forme adaptée.
L’estimateur ainsi obtenu pour le risque espéré est asymptotiquement (lorsque \(k = N\) tend vers l’infini) sans biais. Pour \(k = N\) fini, cet estimateur présente néanmoins un léger biais : il surestime le risque espéré car l’apprentissage se fait à chaque fois sur moins de \(N\) données. Le biais sera ainsi plus faible pour leave one out que pour k-fold. La variance de l’estimateur reste relativement élevée, en partie à cause de l’utilisation de relativement peu de données pour évaluer chaque modèle. On considère que la validation croisée présente un bon compromis entre le biais et la variance de l’estimateur.
Une lecture attentive aura permis de remarquer que, pour répondre à l’objectif d’estimation du risque espéré pour un modèle, nous avons choisi d’en développer (et évaluer) \(k\). Pour lequel de ces modèles est valide cette estimation du risque espéré ? En général, afin de traiter un problème de modélisation, avec un même ensemble \(\mathcal{D}_N\) sont employées plusieurs « procédures » de modélisation, chacune définie par une famille \(\mathcal{F}\), un critère de régularisation \(G(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc. La validation croisée sert ensuite à choisir, entre ces « procédures » de modélisation, celle pour laquelle le risque espéré estimé a la valeur la plus faible. La variance de l’estimateur restant relativement élevée, ce choix n’est pas nécessairement optimal. La « procédure » ainsi choisie est enfin employée pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). C’est à ce modèle que s’applique l’estimation du risque espéré obtenue pour la « procédure » dont le modèle est le résultat. Nous reviendrons plus loin sur les questions de sélection de modèle (ou de « procédure » de modélisation).
L’utilisation de la validation croisée impose, dans certains cas, des précautions :
Pour les problèmes de classement avec des classes déséquilibrées (certaines classes sont bien plus rares que d’autres) : afin d’être assuré de conserver les rapports entre les classes dans tous les découpages, il est nécessaire d’utiliser
Un partitionnement adapté pour la méthode k-fold, par exemple StratifiedKFold dans Scikit-learn.
Un échantillonnage stratifié pour la méthode shuffle and split, par exemple StratifiedShuffleSplit dans Scikit-learn.
La méthode leave one out (LOO) peut être employée telle quelle.
Pour les problèmes dans lesquels les observations ne sont pas indépendantes :
Dans le cas des séries temporelles, les observations successives sont corrélées. Le découpage doit alors être fait par séquences (permettant de conserver un historique local) sur les observations ordonnées et non après shuffle sur les observations individuelles.
Dans le cas des données groupées, à l’intérieur d’un même groupe les observations ne sont pas indépendantes. Les données de test doivent alors provenir de groupes différents de ceux dont sont issues les données d’apprentissage.
L’évaluation de base d’un modèle de classement (ou modèle discriminant) sur un ensemble de données \(\mathcal{E}\) est réalisée à travers le taux de mauvais classement des données de l’ensemble de test, qui correspond à l’erreur moyenne (sur les données de test) obtenue en utilisant la perte 0-1. Une telle évaluation est toutefois réductrice car la perte 0-1 fait l’hypothèse d’un coût de mauvais classement symétrique : le coût est égal à 1 quelle que soit le sens de l’erreur d’affectation. Or, dans de nombreux cas les erreurs d’affectation ne sont pas équivalentes. Par exemple, ne pas détecter la maladie grave d’un patient à partir de mesures cliniques est dramatique (observation de la classe 1 affectée par erreur à la classe 0), alors que signaler une maladie grave pour un patient qui n’en est pas atteint oblige simplement à effectuer un ou plusieurs examens complémentaires (observation de la classe 0 affectée par erreur à la classe 1). Un autre exemple : pour un cargo, la non détection d’un autre navire par le radar peut mener à une collision, alors qu’une fausse alerte provoque seulement un ralentissement temporaire. Bien entendu, si cette asymétrie du coût peut être quantifiée alors elle peut être prise en compte dès le départ, dans la construction du modèle décisionnel. Il est toutefois utile de pouvoir comparer plusieurs modèles discriminants sans fixer a priori un « degré » d’asymétrie.
Les courbes ROC (initiales provenant de receiver operating characteristic, ou « caractéristique de fonctionnement du récepteur » en traduction littérale) répondent à cet objectif. On considère un problème de discrimination dans lequel une des classes est la « classe d’intérêt » et un modèle de classement (ou discriminant) appris à partir des données est vu comme le « détecteur » de la classe d’intérêt.
Un modèle discriminant est en général décrit par un vecteur de paramètres (par ex. \(\mathbf{w}\), qui indique l’orientation de l’hyperplan de séparation pour un modèle linéaire) et un seuil de détection (\(b\), la position de l’hyperplan pour le modèle linéaire). Le principe d’une courbe ROC est de balayer la totalité du domaine utile de variation de ce seuil et d’examiner, pour chaque valeur du seuil, les résultats obtenus sur les données d’évaluation, représentés par la matrice de confusion :
Classe présente
Classe absente
Classe détectée
Vrai Positif
Faux Positif
Classe non détectée
Faux Négatif
Vrai Négatif
Chaque donnée de \(\mathcal{E}\) peut appartenir à la classe d’intérêt (Classe présente) ou à une autre classe (Classe [d’intérêt] absente) ; cette information est supposée connue pour toutes les données de \(\mathcal{E}\) (« vérité terrain »). Chaque donnée peut être affectée par le modèle à la classe d’intérêt (Classe détectée) ou à une autre classe (Classe non détectée). Si une donnée fait partie de la classe d’intérêt mais est affectée par le modèle à une autre classe, on parle d’un « faux négatif ». Si une donnée est affectée par le modèle à la classe d’intérêt alors qu’elle fait partie d’une autre classe, on parle d’un « faux positif ».
A partir du contenu d’une matrice de confusion, on définit les mesures suivantes :
La sensibilité, ou le taux de vrais positifs, mesure la capacité du modèle à détecter les vrais positifs. Si tous les vrais positifs sont détectés (s’il n’y a pas de faux négatifs, c’est à dire des positifs non détectés comme positifs), alors la sensibilité est égale à 1.
La 1 - spécificité, ou le taux de faux positifs, mesure la capacité du modèle à détecter seulement les vrais positifs. Si aucun négatif n’est détecté comme positif (s’il n’y a pas de faux positifs), alors la 1 - spécificité est égale à 0.
Dans la figure suivante sont représentées des données de \(\mathbb{R}^2\). Celles de la classe d’intérêt sont en vert et celles des autres classes en rouge. Considérons trois modèles discriminants qui sont des PMC avec \(\alpha = 1\) correspondant à trois valeurs différentes du seuil. Chaque modèle discriminant est une frontière de séparation non linéaire. Dans cet exemple, la classe d’intérêt n’est jamais parfaitement séparée des autres classes, quelle que soit la position de la frontière.
Fig. 27 Frontières de discrimination pour 3 valeurs du seuil de détection¶
Une courbe ROC montre le taux de vrais positifs (en ordonnée) en fonction du taux de faux positifs (en abscisse), la variable étant le seuil. Pour le problème illustré dans la figure précédente, en explorant la totalité du domaine de variation utile du seuil et en enregistrant à chaque fois la sensibilité (ou taux de vrais positifs) et 1 - spécificité (ou taux de faux positifs), on obtient le graphique suivant :
Fig. 28 Courbe ROC associée à l’exemple de la Fig. 27¶
Lorsque la frontière est positionnée très haut dans la Fig. 27, toutes le données sont du côté « négatif ». Aucun vrai positif n’est détecté et aucun négatif n’est détecté comme positif. La sensibilité est donc nulle et la 1 - spécificité est nulle également.
Au fur et à mesure que la frontière se déplace vers le bas et vers la gauche dans la Fig. 27, de plus en plus de positifs sont détectés mais aucun négatif n’est encore détecté comme positif. La sensibilité augmente et la 1 - spécificité reste nulle. A partir d’une valeur du seuil, des négatifs commencent à être détectés comme positifs. La 1 - spécificité commence donc à augmenter. Si la frontière continue son déplacement vers le bas et la gauche, à partir d’une autre valeur du seuil il n’y a plus aucun positif du mauvais côté de la frontière, tous sont détectés comme des positifs. La sensibilité atteint donc 1. Noter que dans l’exemple de la Fig. 27 un des points verts est très éloigné de sa classe, ce fait que la sensibilité n’atteint pas 1 avant que la 1 - spécificité ait atteint 1, comme on peut le voir sur la courbe ROC de la Fig. 28. En revanche, comme de plus en plus de négatifs sont détectés comme positifs, la 1 - spécificité continue à augmenter.
Lorsque la frontière est suffisamment en bas à gauche dans la Fig. 27, toutes le données sont du côté « positif ». Tous les positifs sont détectés et, en plus, tous les négatifs sont détectés comme positifs. La sensibilité est donc égale à 1 mais la 1 - spécificité est égale à 1 aussi. Noter que ce cas n’est pas illustré sur la Fig. 28 car on s’arrête lorsque la 1 - spécificité atteint 1 même si la sensibilité est encore inférieure à 1.
Peut-on comparer ces modèles à partir de leurs courbes ROC, pour constater éventuellement qu’un est plus intéressant (ou moins intéressant) que les autres sur la totalité du domaine de variation du seuil ? Comparons les résultats avec ceux de deux autres modèles, un modèle linéaire (obtenu par AFD) et un autre qui est un PMC avec \(\alpha = 10^{-5}\). La figure suivante montre les frontières de discrimination obtenues par chaque modèle pour trois valeurs différentes du seuil, ainsi que les courbes ROC correspondantes :
Fig. 29 Comparaison de modèles à travers leurs courbes ROC et les aires sous la courbe¶
Les trois courbes ROC regroupées sur un même graphique sont montrées dans la figure suivante. La courbe ROC du PMC \(\alpha = 1\) se situe presque systématiquement au-dessus des deux autres, en revanche les courbes ROC des deux autres modèles se croisent plusieurs fois. Une mesure couramment employée pour comparer deux modèles sur la totalité du domaine de variation utile du seuil est l”aire sous la courbe ROC, notée en général AUC (area under curve). Plus sa valeur est élevée, plus le modèle est intéressant (performant). Si deux modèles ont des valeurs AUC très proches, le choix doit être fait à partir d’autres critères, par exemple en comparant les sensibilités à spécificité fixée.
Fig. 30 Courbes ROC : modèle linéaire en bleu, PMC \(\alpha = 10^{-5}\) en rouge, PMC \(\alpha = 1\) en vert¶
Le traitement d’un problème de modélisation décisionnelle à partir d’un ensemble de données \(\mathcal{D}_N\) passe en général par l’évaluation de plusieurs « procédures » de modélisation différentes, chacune définie par une famille de modèles \(\mathcal{F}\), des paramètres spécifiques à la famille choisie (par ex. l’architecture pour un PMC, le type et la variance du noyau pour une SVM), un critère de régularisation \(G(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc.
Les paramètres numériques dont dépend une procédure de modélisation sont en général appelés « hyperparamètres », bien que ce terme ait été employé à l’origine pour les paramètres des distributions a priori dans les statistiques bayesiennes. Par exemple, pour les SVM à noyau RBF sont considérés hyperparamètres la variance du noyau RBF et la valeur de la constante \(C\) de régularisation. Parfois, le terme « hyperparamètres » est étendu aux variables nominales définissant le famille de modèles \(\mathcal{F}\), comme par exemple le type de noyau employé pour des SVM.
Le différentes « procédures » de modélisation sont ensuite comparées à travers leurs scores de validation croisée afin de retenir celle pour laquelle le risque espéré (l’erreur de généralisation) présente la valeur la plus faible. La « procédure » ainsi choisie est enfin employée pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). Une fois trouvé le « meilleur » modèle (par la procédure de modélisation retenue), son risque espéré doit être estimé sur des données de qui n’ont servi ni à la recherche des paramètres, ni à celle des hyperparamètres !
Pour trouver le meilleur modèle décisionnel il est nécessaire de comparer plusieurs « procédures » de modélisation différentes. Chaque « procédure » de modélisation est définie par une valeur spécifique pour chaque hyperparamètre. Une fois fixées les valeurs des hyperparamètres (par ex. de \(C\) pour les SVM linéaires), l’algorithme d’optimisation (par ex. la descente de sous-gradient stochastique) opère pour déterminer le modèle (par ex. \(\mathbf{w}^*\) et \(b^*\) pour une SVM linéaire).
Les meilleures valeurs des hyperparamètres sont identifiées de la manière suivante :
Définition d’ensembles de valeurs pour les éventuels hyperparamètres nominaux (par ex. architectures PMC, critères de régularisation, types de noyaux pour les SVM).
Définition d’intervalles de variation pour les hyperparamètres numériques et d’une procédure d’exploration de cet espace.
Exploration de l’espace des hyperparamètres suivant la procédure choisie. Pour chaque tuple de valeurs (une valeur pour chaque hyperparamètre), apprentissage et évaluation des modèles résultants par application de la validation croisée.
Comparaison des résultats de validation croisée, sélection des valeurs des hyperparamètres qui mènent aux meilleures performances (à l’erreur de généralisation estimée la plus faible).
Une procédure d’exploration simple est la recherche systématique dans une grille (grid search) : si on dispose de \(m\) hyperparamètres numériques, on définit pour chaque hyperparamètre un intervalle et un pas de variation, on obtient ainsi une grille (de dimension \(m\)) dont les nœuds correspondent aux valeurs à tester. Pour chaque nœud de la grille (tuple de \(m\) valeurs), la validation croisée est appliquée afin d’obtenir une estimation de l’erreur de généralisation des modèles correspondants. Bien entendu, tous les nœuds de la grille peuvent être explorés en parallèle si une plate-forme adéquate est disponible.
Avec une grille trop fine, le nombre de modèles à explorer peut être excessif. Avec une grille trop grossière, l’exploration de l’espace des paramètres peut être trop grossière et ne pas permettre de trouver les meilleurs modèles. Une solution souvent adoptée est celle d’une grille qui a plusieurs niveaux de « finesse », comme dans la figure suivante. Dans un tel cas, la recherche est hiérarchique : sont d’abord explorées les valeurs correspondant à la grille grossière, ensuite des grilles plus fines sont définies dans les intervalles de la grille grossière où les meilleurs résultats ont été trouvés, et sont explorées enfin les valeurs issues de ces grilles plus fines. Cette exploration hiérarchique permet d’obtenir une exploration plus fine à moindre coût qu’une recherche exhaustive au niveau le plus fin. Le procédé peut être répété à plusieurs niveaux. A chaque niveau, les nœuds de la grille peuvent être explorés en parallèle.
Fig. 31 Grille à deux niveaux pour recherche hiérarchique¶
Il est important de noter que pour des méthodes d’apprentissage spécifiques il est parfois possible de définir des procédures d’exploration plus efficaces.
Les valeurs des hyperparamètres qui mènent à l’erreur de généralisation estimée la plus faible sont retenues et employées pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). C’est ce modèle qui est retourné par la procédure de recherche en grille.
Enfin, pour estimer le risque espéré (l’erreur de généralisation) du modèle retenu in fine il faut employer des données qui n’ont été utilisées ni pour l’apprentissage du modèle (pour l’algorithme d’optimisation des paramètres), ni pour la recherche des meilleures valeurs des hyperparamètres !
Lorsque des connaissances a priori permettent de privilégier certains intervalles de variation pour les hyperparamètres, la recherche aléatoire à partir de valeurs générées en conformité avec ces connaissances présente une meilleure efficacité qu’une grid search non hiérarchique.
Concrètement, pour chaque hyperparamètre une loi de tirage est définie, ensuite des tuples de valeurs (une valeur pour chaque hyperparamètre) sont obtenues et un modèle est appris avec ce tuple de valeurs. Les échantillons sont générés en considérant en général les hyperparamètres comme étant indépendants. Un intérêt particulier de cette approche est que le coût peut être maîtrisé en fixant le nombre d’échantillons à générer.
Les modalités d’échantillonnage dépendent de la nature des hyperparamètres considérés :
pour les hyperparamètres numériques à valeurs continues (par ex. la pondération \(\alpha\) de la régularisation) on précise la loi d’échantillonnage, par ex. loi gamma de paramètres donnés ;
pour les hyperparamètres numériques à valeurs discrètes (par ex. nombre de neurones cachés) on précise la loi d’échantillonnage, par ex. loi uniforme sur un intervalle donné ;
pour les hyperparamètres variables nominales, on précise la liste des valeurs (modalités) possibles et on considère une loi uniforme sur ces valeurs.
Évaluation et sélection de modèles : que faut-il retenir ?¶
Le risque espéré (l’erreur de généralisation) doit être estimé sur des données non utilisées pour l’apprentissage.
La validation croisée permet une estimation de plus faible variance qu’un seul découpage apprentissage | test, ainsi qu’une meilleure utilisation des données disponibles.
Le courbes ROC facilitent une comparaison plus globale de modèles de classement (ou de discrimination).
Pour obtenir les meilleures valeurs pour les hyperparamètres on emploie une recherche systématique ou aléatoire, la comparaison des modèles étant réalisée par validation croisée.
Si la validation croisée est employée pour sélectionner le meilleur modèle, l’estimation du risque espéré du modèle retenu doit être faite sur des données non encore utilisées.
O. Bousquet, S. Boucheron, and G. Lugosi. Introduction to Statistical Learning Theory, pages 169–207. Volume Lecture Notes in Artificial Intelligence 3176. Springer, Heidelberg, Germany, 2004.
Olivier Chapelle, Bernhard Schölkopf, and Alexander Zien, editors. Semi-Supervised Learning. MIT Press, Cambridge, MA, 2006. URL: http://www.kyb.tuebingen.mpg.de/ssl-book.
Lixuan Yang, Helena Rodriguez, Michel Crucianu, and Marin Ferecatu. Fully convolutional network with superpixel parsing for fashion web image segmentation. In Proc. 23rd Intl. Conf. MultiMedia Modeling, Reykjavik, Iceland, 139–151. 2017.
 ?
?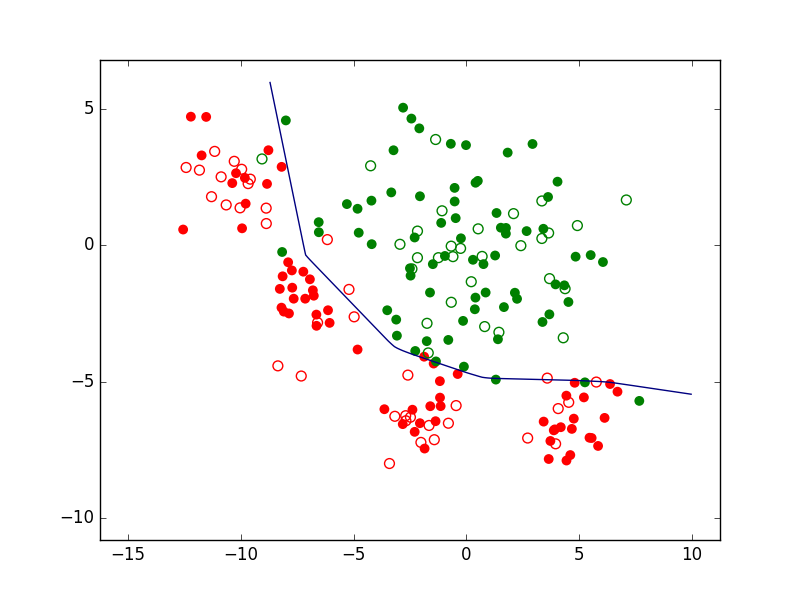
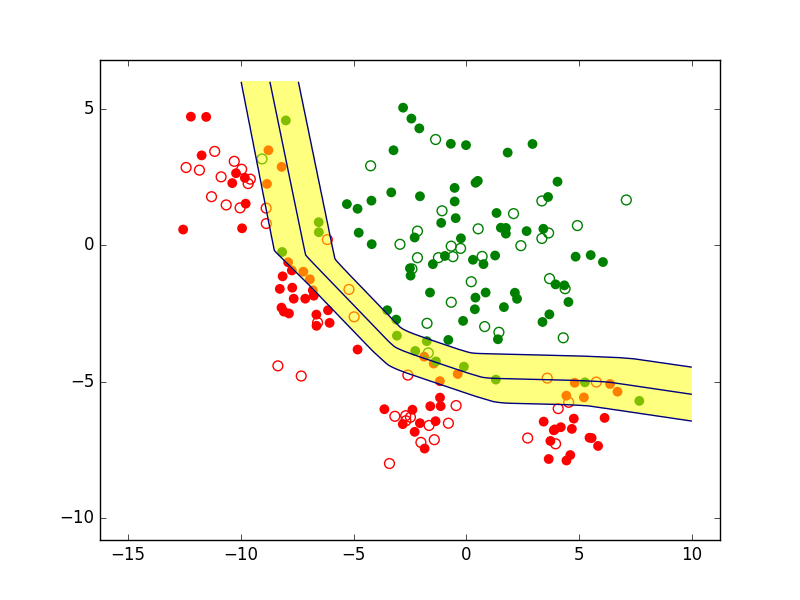
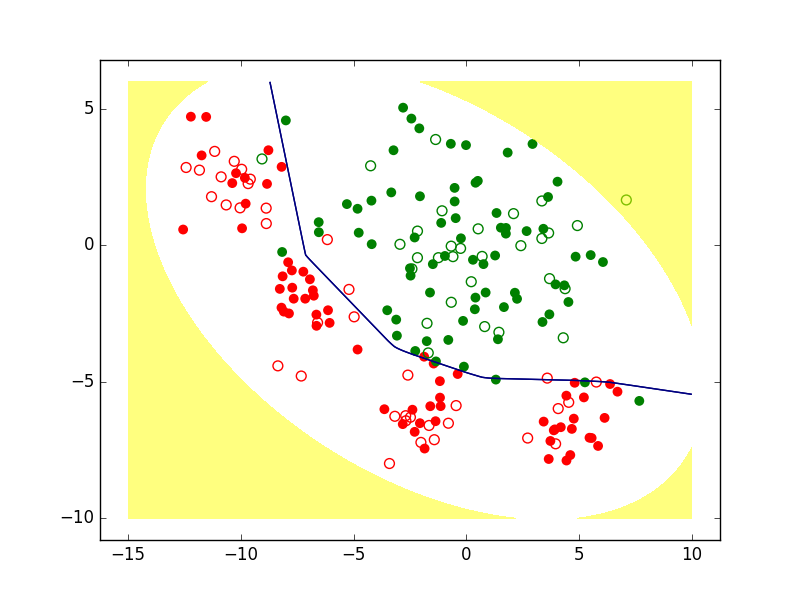
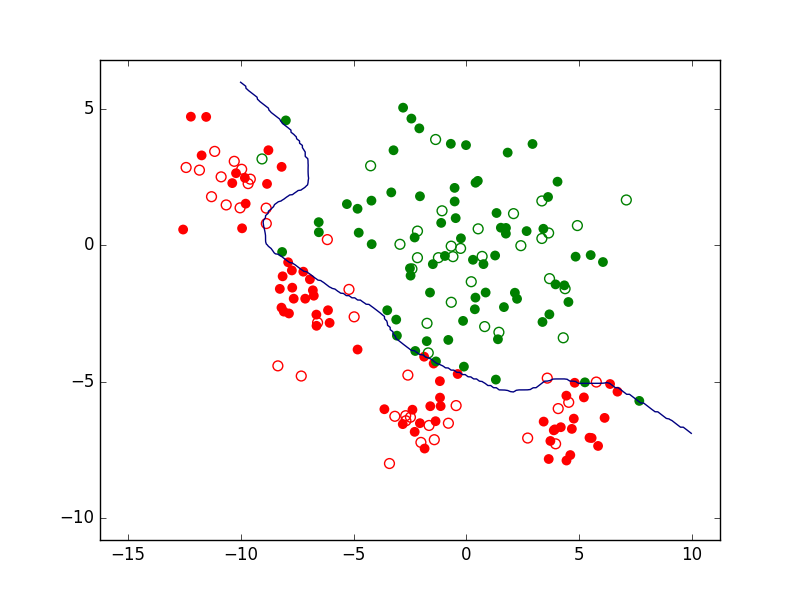
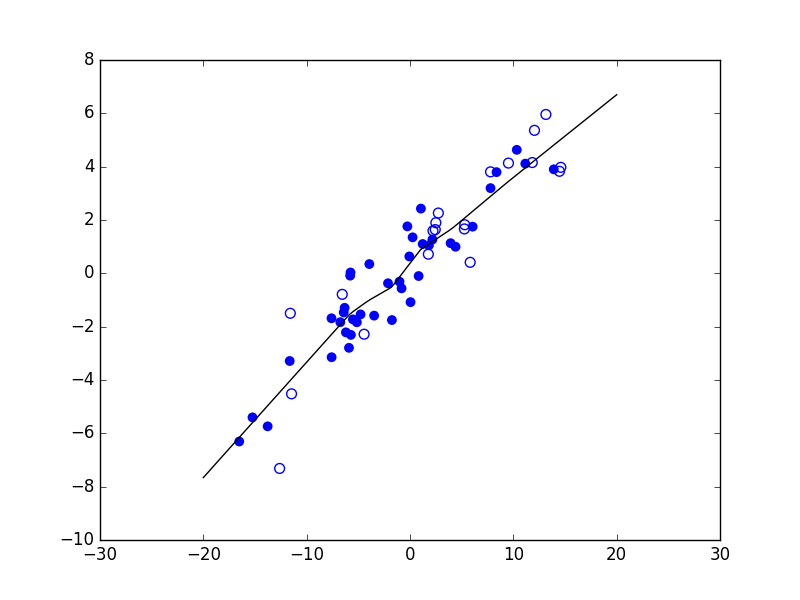
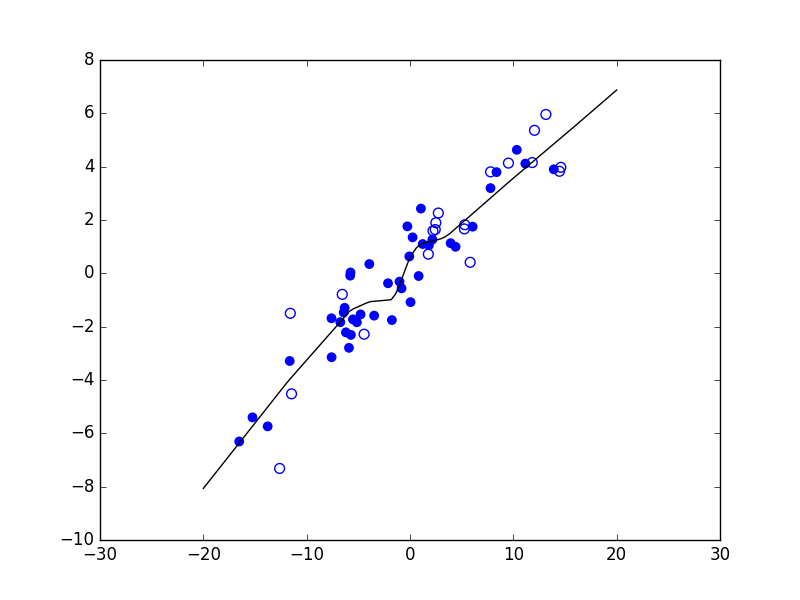

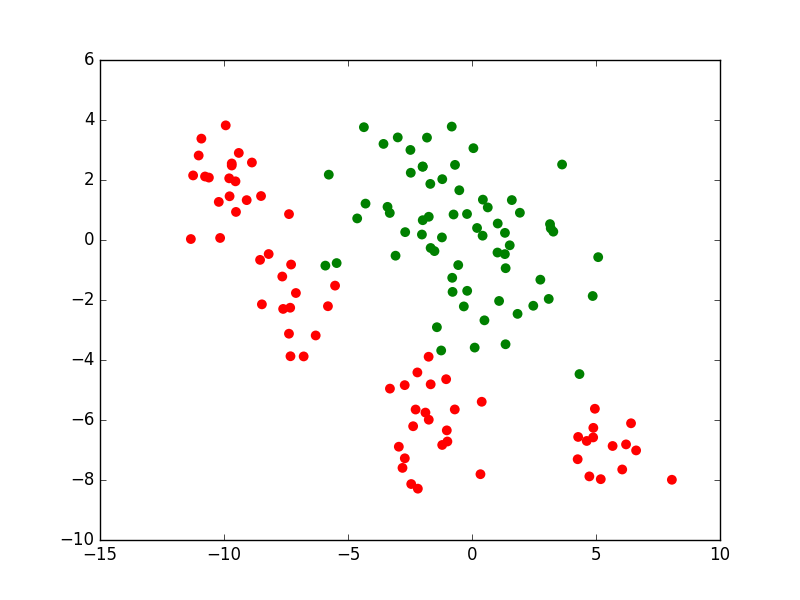
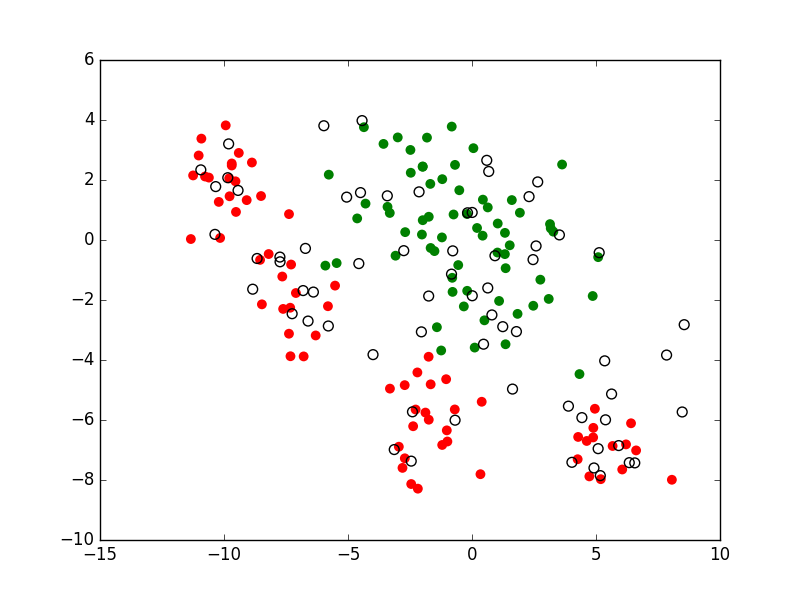
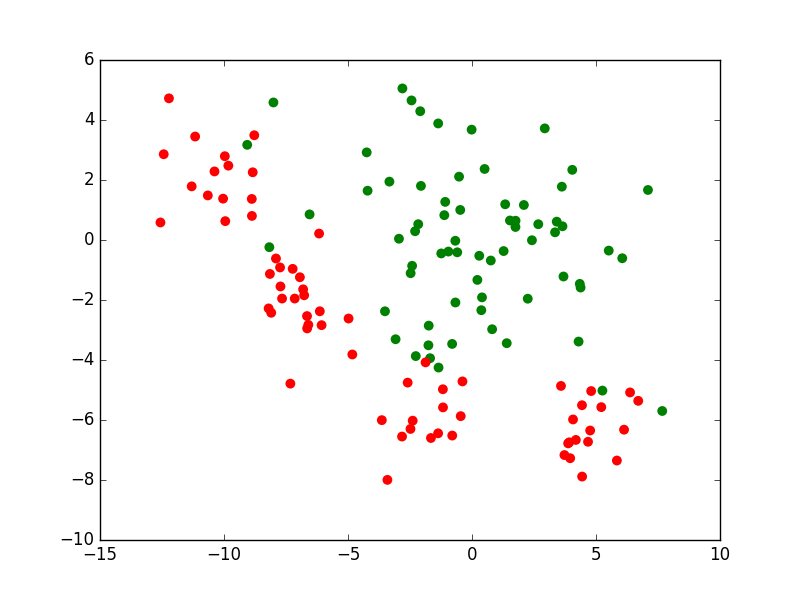
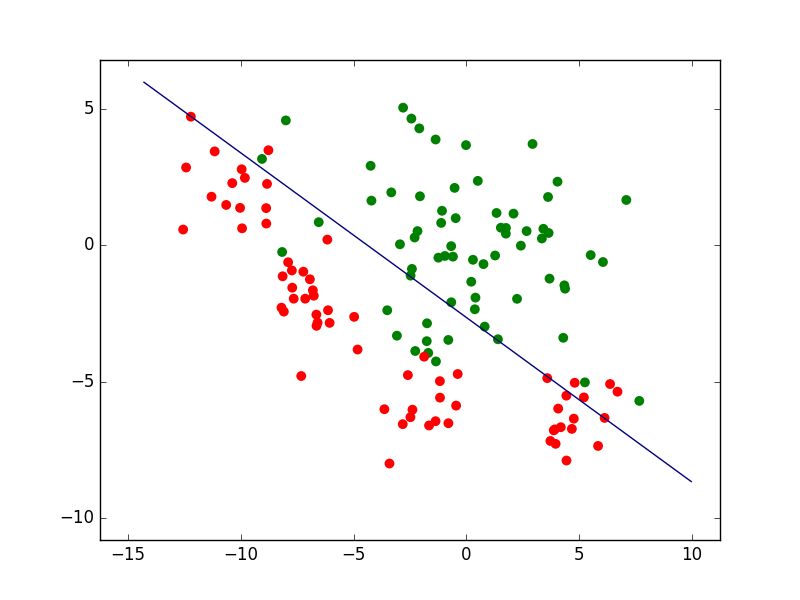
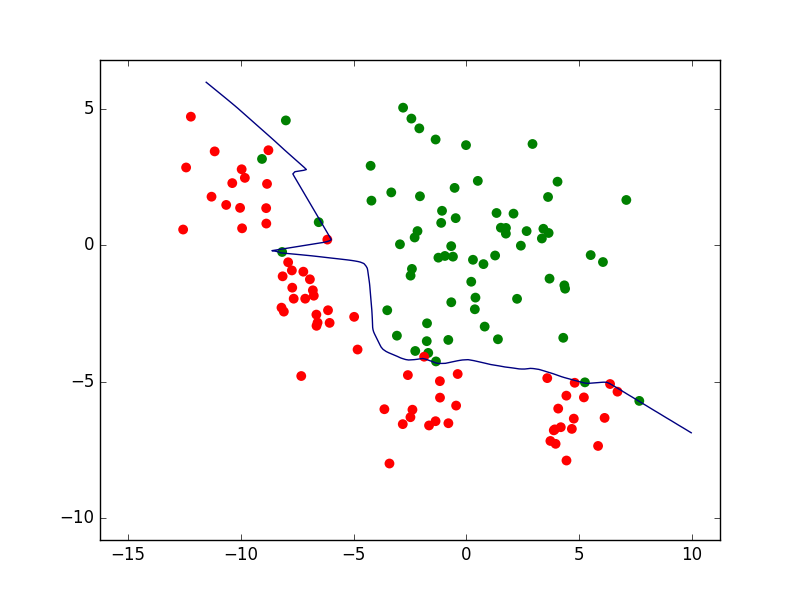
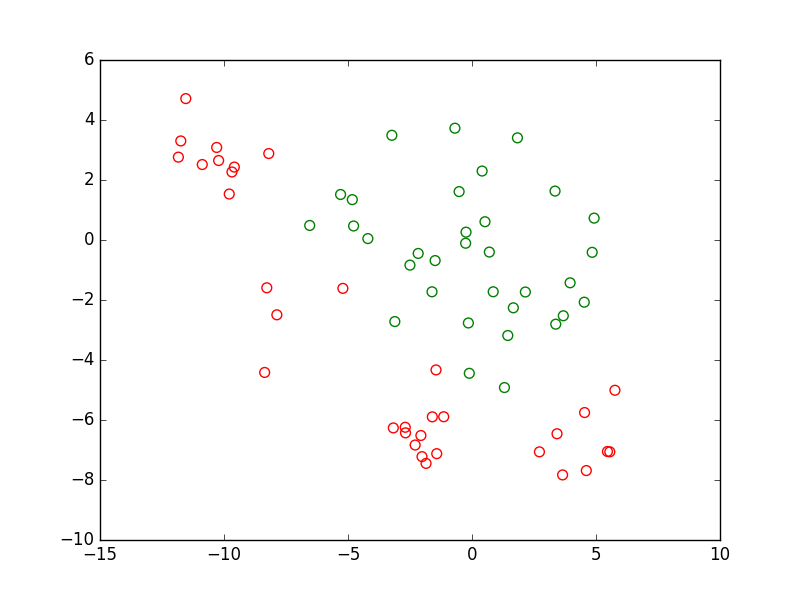
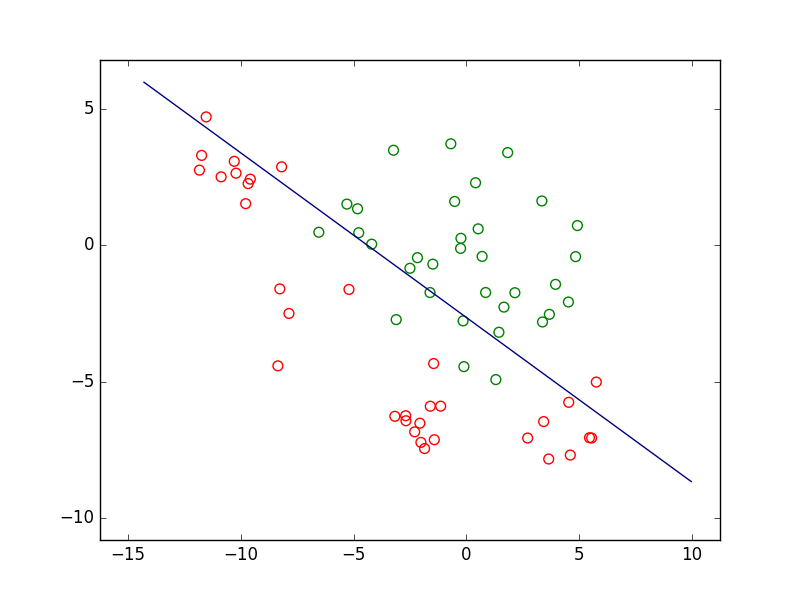
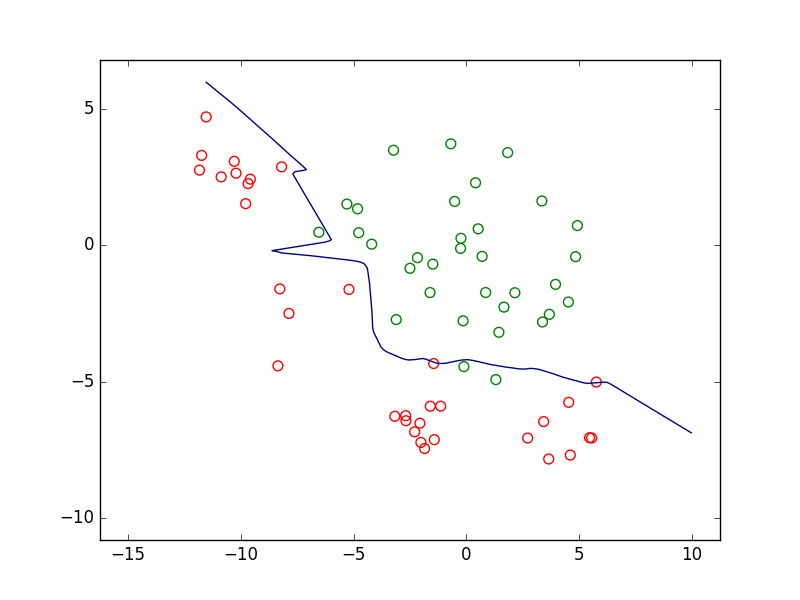
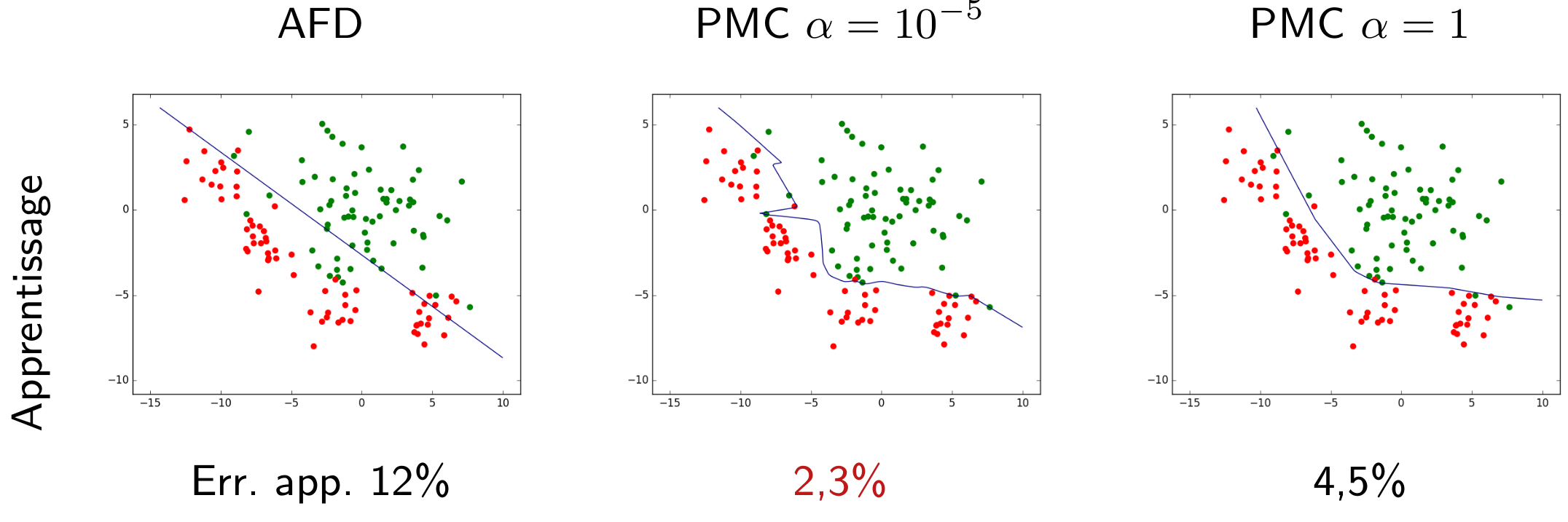
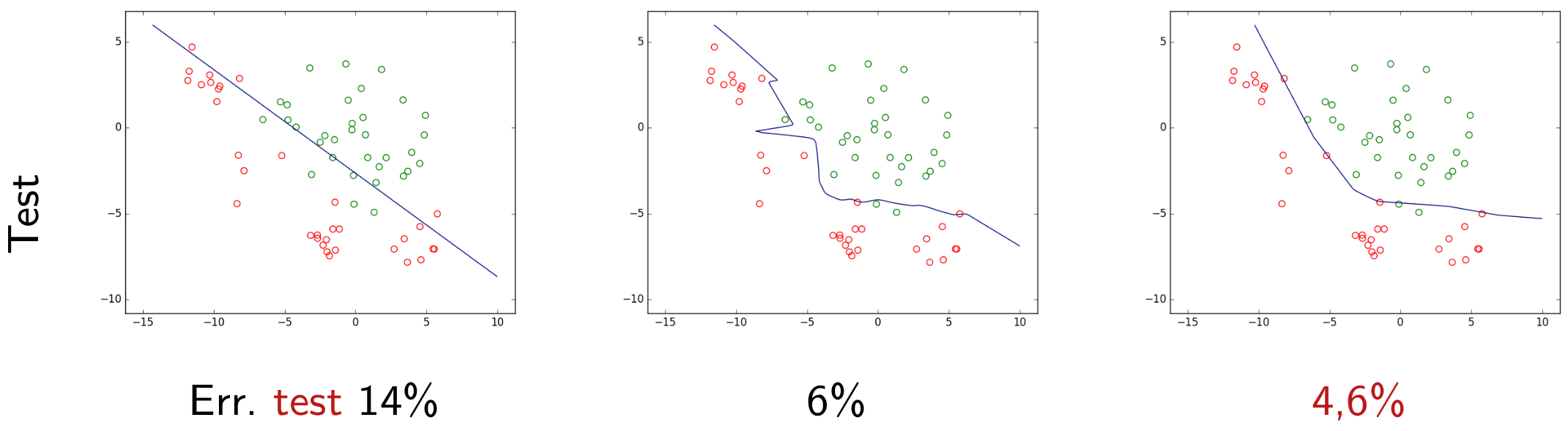
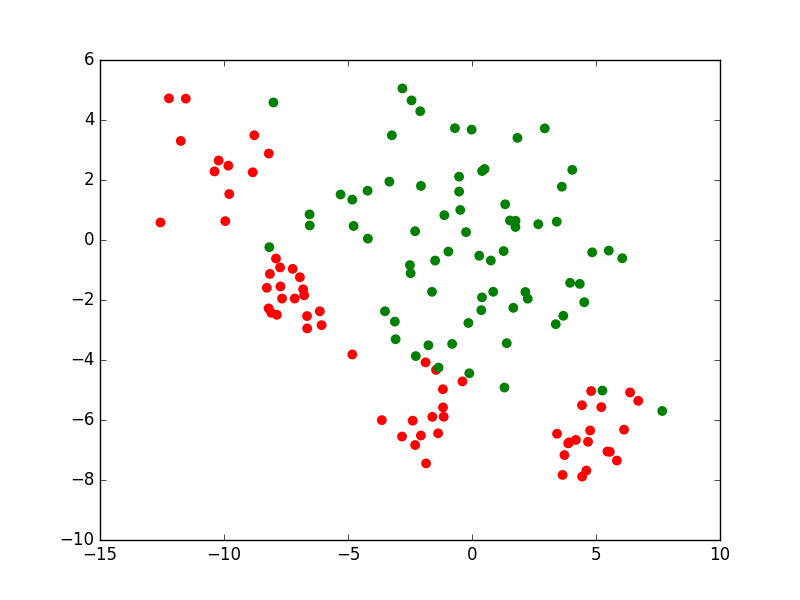
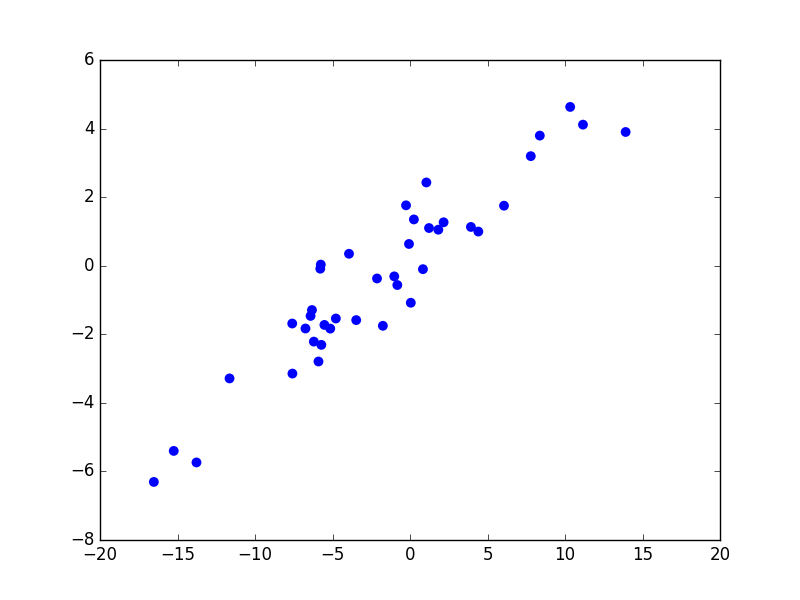
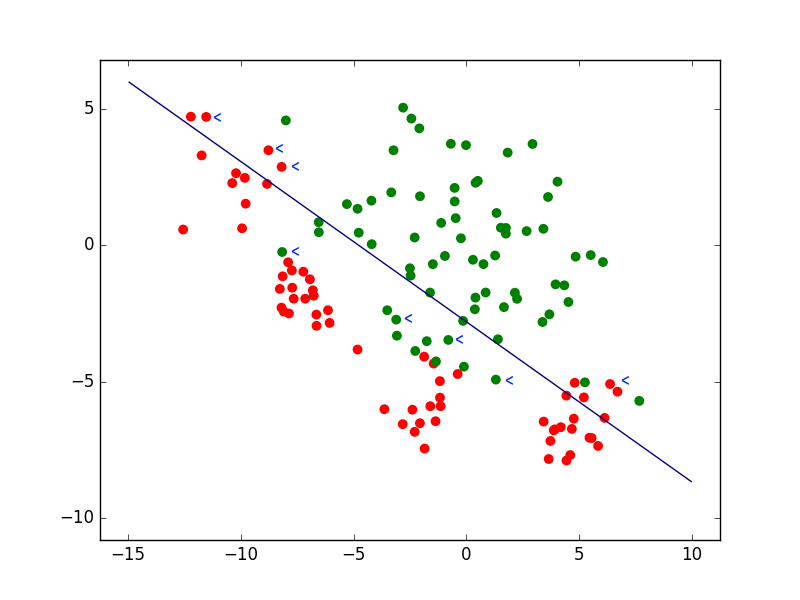
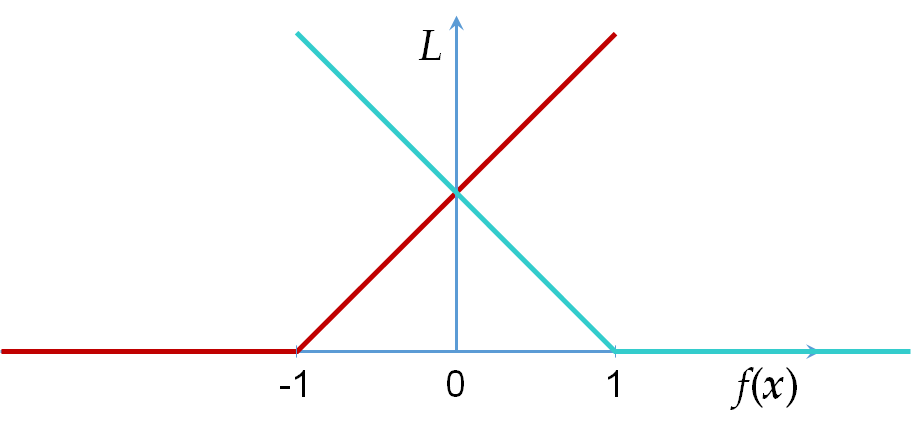
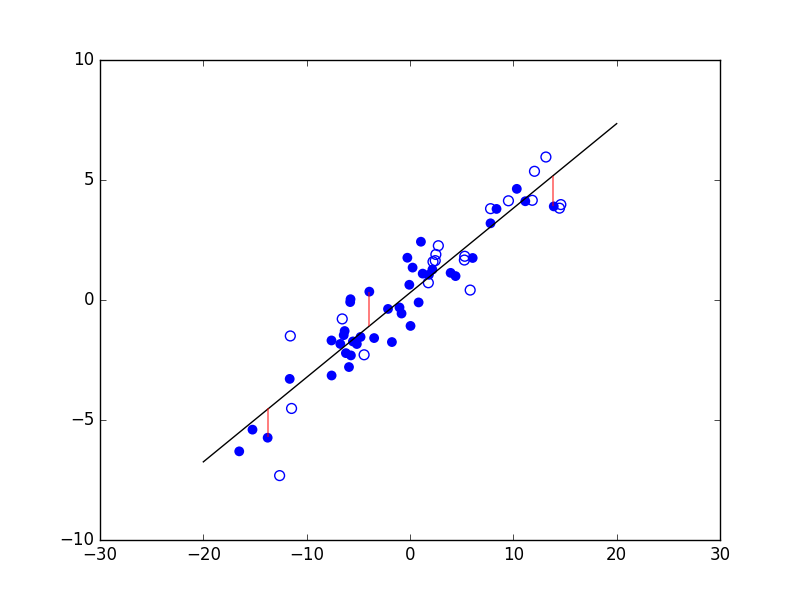
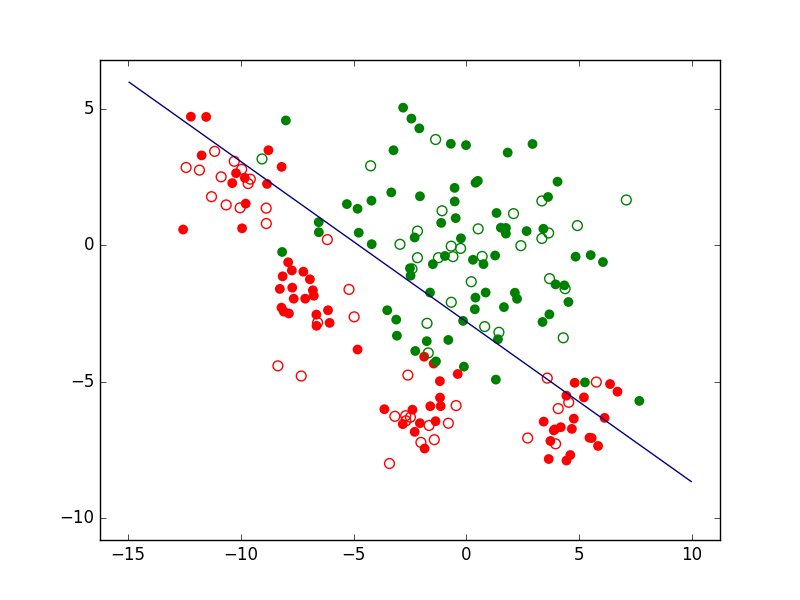
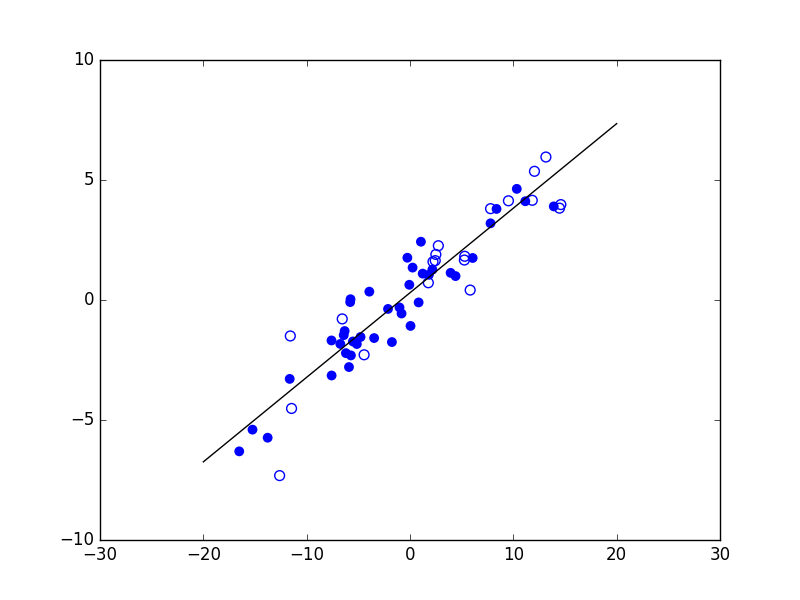
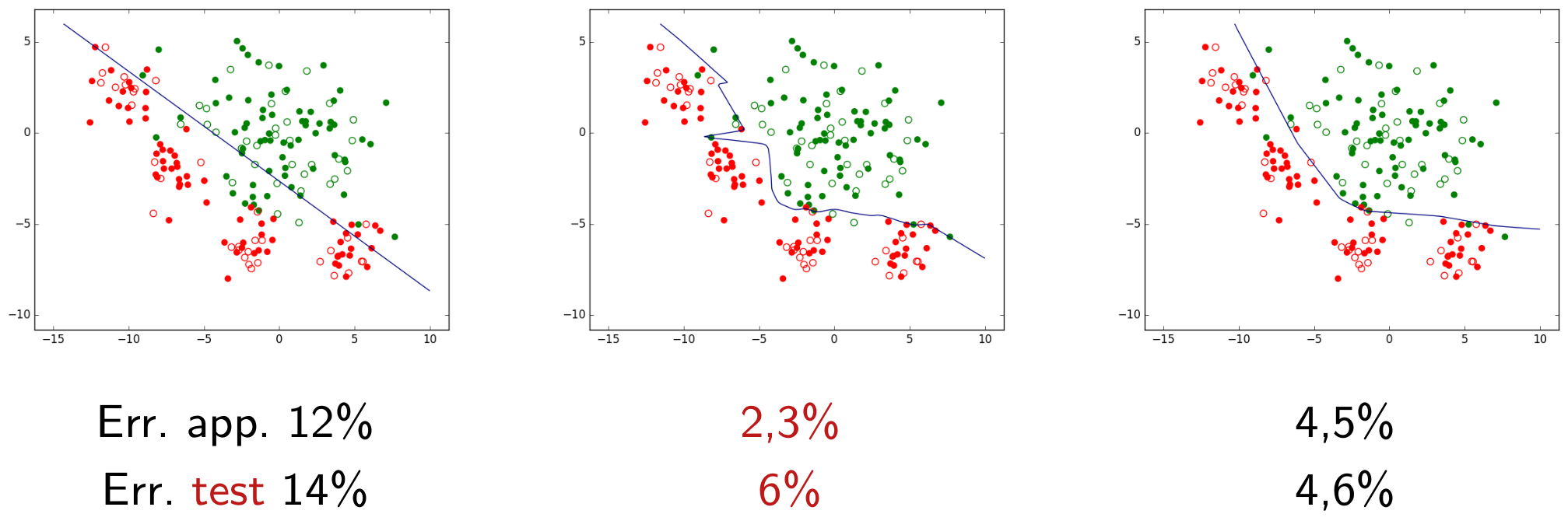
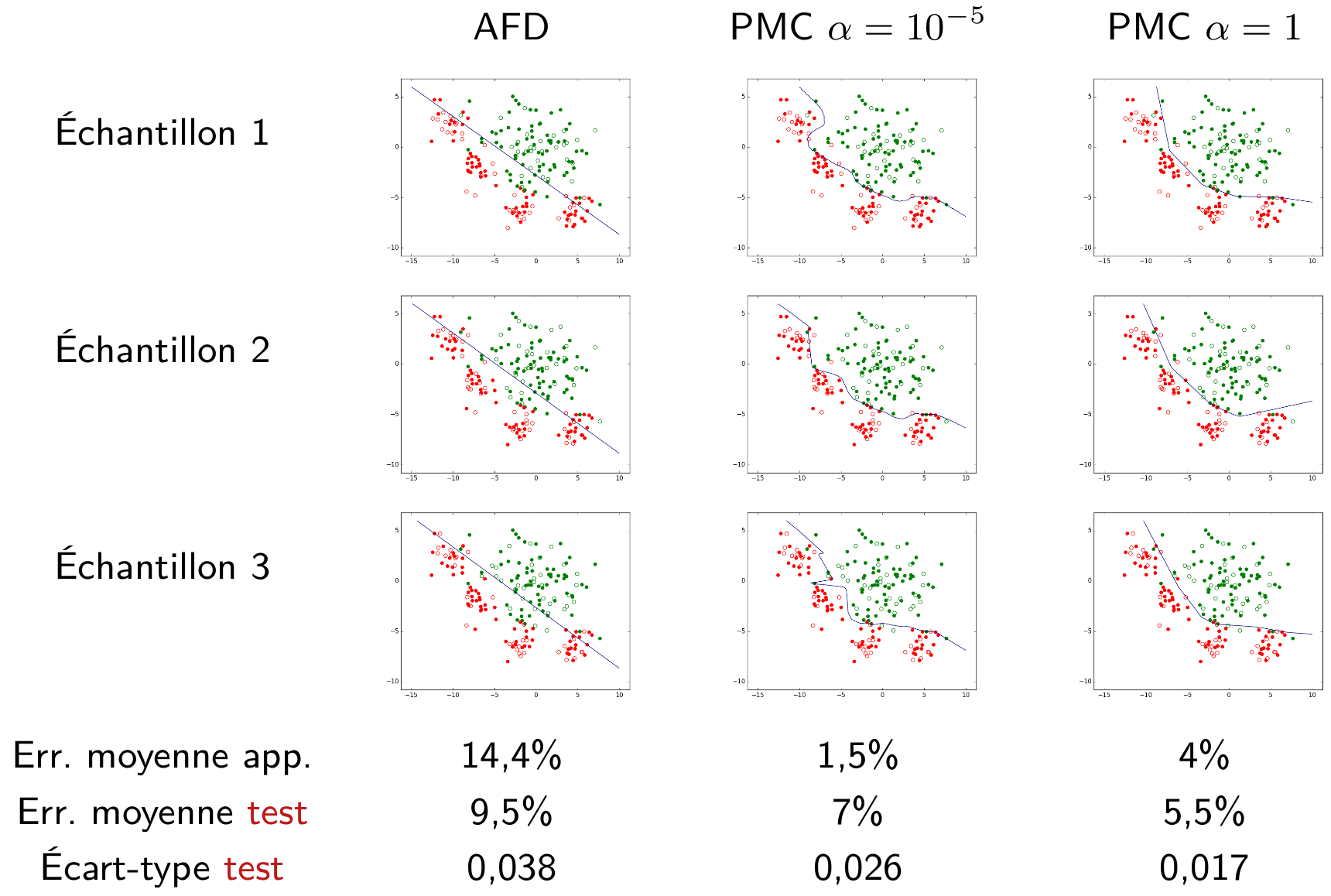


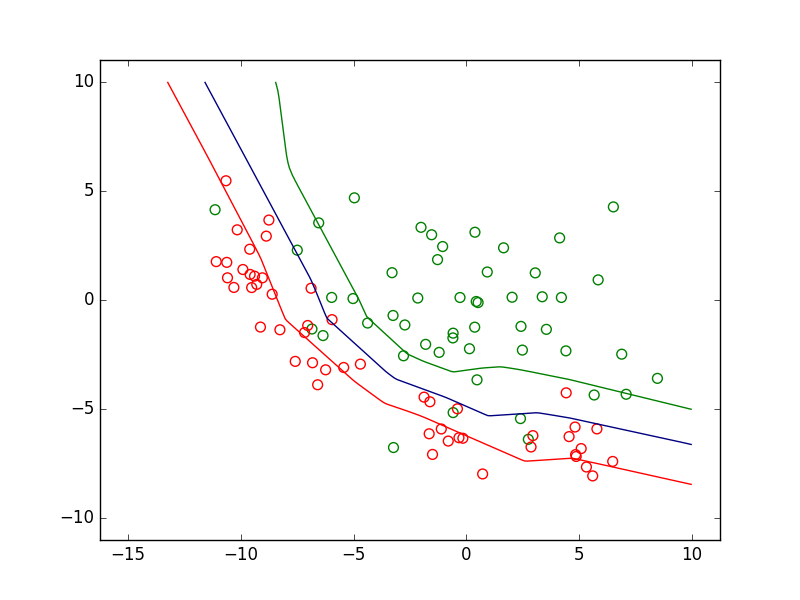
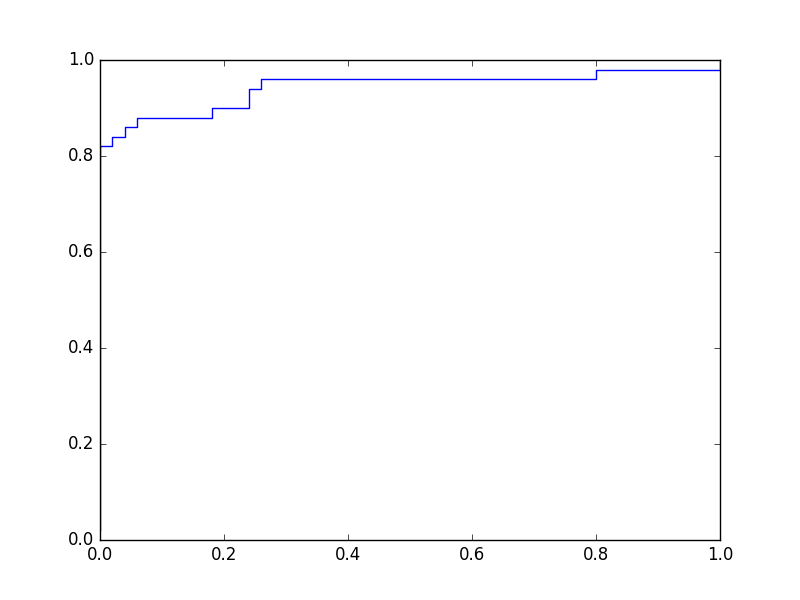
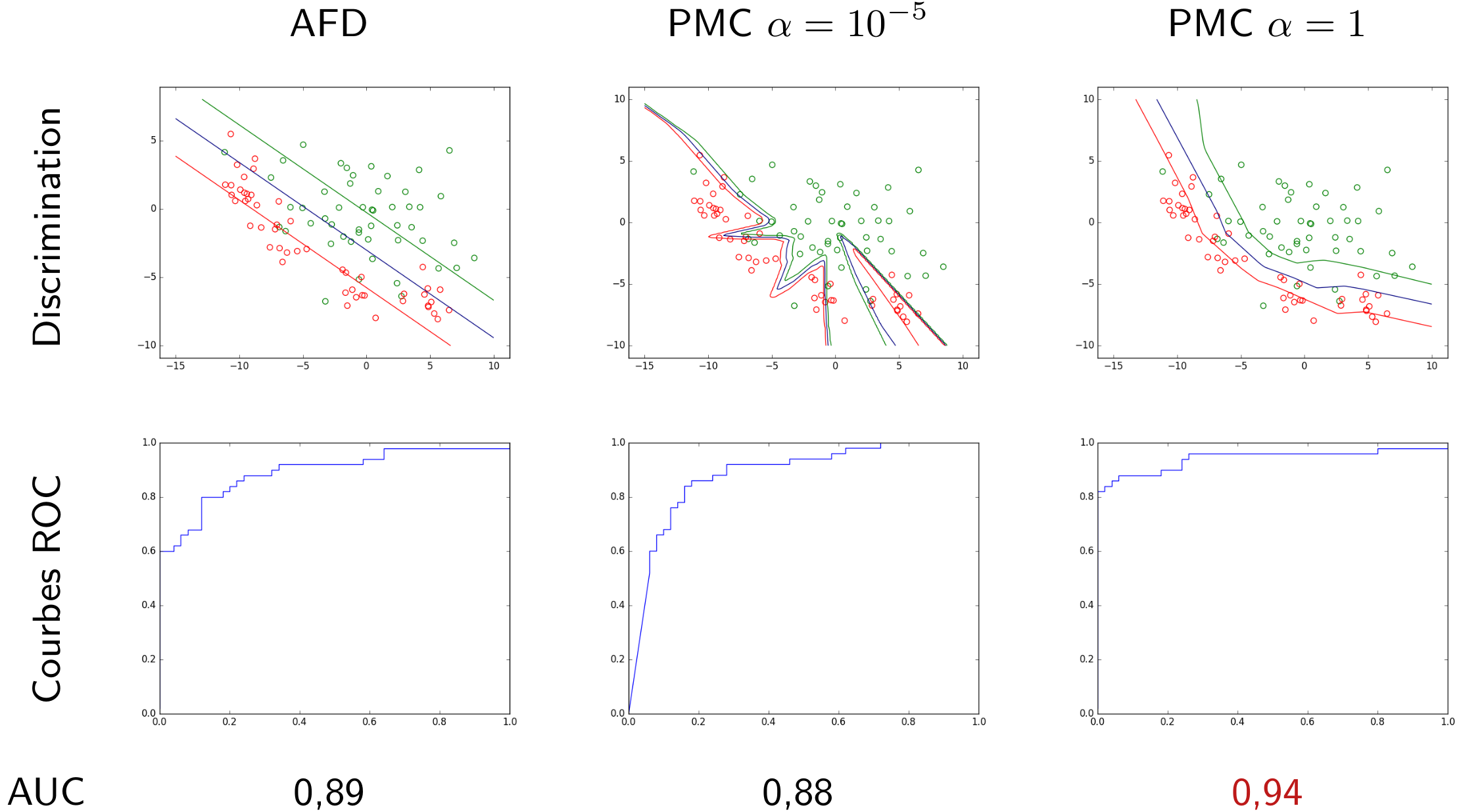
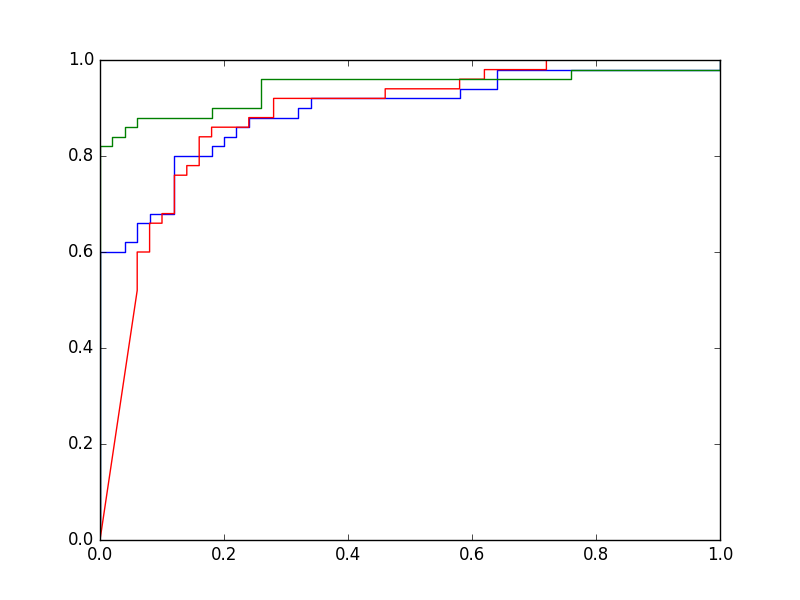
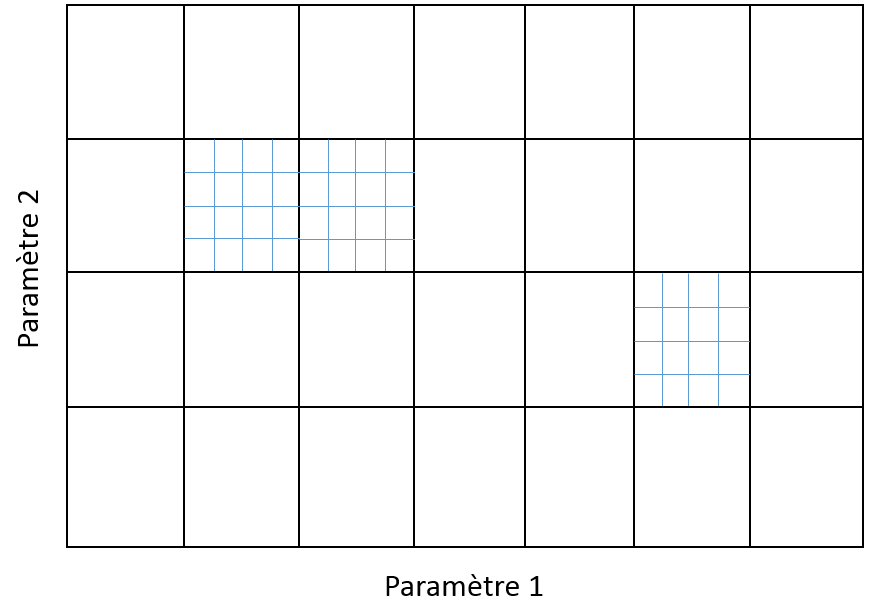
Comment mesurer la capacité ?¶
Jusqu’ici nous avons parlé de la capacité (d’approximation) d’une famille de modèles de façon intuitive, sans définir une mesure précise. Nous présentons brièvement dans cette section la mesure de capacité la plus connue, la dimension de Vapnik-Chervonenkis, qui permet d’obtenir plusieurs résultats intéressants (voir aussi le chapitre sur les SVM).
Pour introduire cette mesure, considérons un ensemble de \(N\) vecteurs \(\{\mathbf{x}_i\}_{1 \leq i \leq N} \in \mathbb{R}^p\) (vecteurs à composantes réelles, de dimension \(p\)). Il y a \(2^N\) façons différentes de séparer cet ensemble en deux parties (attention, l’ensemble vide et l’ensemble complet sont aussi des parties). Nous pouvons maintenant évaluer la capacité d’une famille \(\mathcal{F}\) de fonctions \(f : \mathbb{R}^p \rightarrow \{-1, 1\}\) en examinant ses capacités à séparer différents ensembles de vecteurs. Une séparation est « matérialisée » par le fait qu’une fonction \(f \in \mathcal{F}\) associe la valeur \(-1\) à certains vecteurs et la valeur \(1\) aux autres.
Définition : la famille \(\mathcal{F}\) de fonctions \(f : \mathbb{R}^p \rightarrow \{-1, 1\}\) pulvérise \(\{\mathbf{x}_i\}_{1 \leq i \leq N}\) si toutes les \(2^N\) séparations peuvent être construites avec des fonctions de \(\mathcal{F}\).
Définition : l’ensemble \(\mathcal{F}\) est de VC-dimension (dimension de Vapnik-Chervonenkis) \(h\) s’il pulvérise au moins un ensemble de \(h\) vecteurs et aucun ensemble de \(h+1\) vecteurs.
Par exemple, la VC-dimension de l’ensemble des hyperplans (les séparations linéaires de dimension \(p-1\)) de \(\mathbb{R}^p\) est \(h = p+1\). Pour illustrer, considérons le cas particulier de l’espace bidimensionnel \(\mathbb{R}^2\). Dans \(\mathbb{R}^2\) les hyperplans sont les droites. Dans la figure suivante nous pouvons voir un triplet et un quadruplet de points (vecteurs de \(\mathbb{R}^2\)).
L’ensemble des droites de \(\mathbb{R}^2\) pulvérise le triplet de points à gauche car pour chacune des \(2^3 = 8\) séparations envisageables (\(-1\) associé à tous les points et \(1\) à aucun, \(1\) associé à tous les points et \(-1\) à aucun, \(-1\) associé au point de gauche et \(1\) aux autres, etc.) il existe au moins une droite qui permet de l’obtenir. En revanche, on peut montrer qu’aucun quadruplet de points ne peut être pulvérisé (il existe au moins une séparation qui ne peut pas être faite avec une droite). Par exemple, pour le quadruplet représenté dans la figure, aucune droite ne peut séparer les points bleus des points rouges.
La VC-dimension est une mesure intéressante de la capacité car elle permet, entre autres, d’obtenir une borne pour l’écart entre le risque théorique (ou risque espéré) et le risque empirique.
Théorème [BBL04] : soit \(R_{\mathcal{D}_N}(f)\) le risque empirique défini par la fonction de perte \(L_{01}(\mathbf{x},y,f) = \mathbf{1}_{f(\mathbf{x}) \neq y}\); si la VC-dimension de \(\mathcal{F}\) est \(h < \infty\) alors pour toute \(f \in \mathcal{F}\), avec une probabilité au mois égale à \(1-\delta\) (\(0 < \delta < 1\)), on a
La forme de \(B(N,\mathcal{F})\) permet de constater que la borne diminue quand le nombre de données d’apprentissage \(N\) augmente. En effet, avec plus de données d’apprentissage il est possible de mieux « contraindre » le modèle appris et de réduire ainsi la composante « estimation » de l’erreur.
La borne a également une valeur plus faible pour une famille \(\mathcal{F}\) de VC-dimension \(h\) plus faible. Avec un nombre fixé de données d’apprentissage il est possible de mieux « contraindre » un modèle moins « flexible ». Malheureusement, une famille avec la VC-dimension \(h\) trop faible ne sera pas capable de faire la séparation attendue, la composante « approximation » de l’erreur sera trop élevée.
Enfin, la borne peut être réduite par l’augmentation de \(\delta\) (l’écart par rapport à une probabilité de 1), c’est à dire par une diminution des « garanties » qu’apporte la borne.
Il est important de remarquer que cette borne \(B(N,\mathcal{F})\) ne fait pas intervenir le nombre de variables (la dimension de l’espace dans lequel se trouvent les observations), ni la loi conjointe (inconnue) \(P\). En ce sens il s’agit d’un résultat dans le pire des cas, intéressant d’un point de vue théorique mais malheureusement peu utile en pratique car pour obtenir une borne utile (de valeur faible) il faudrait en général disposer d’un nombre \(N\) très élevé d’observations pour l’apprentissage.
Nous pouvons faire une synthèse des conséquences de l’existence d’une borne, \(R(f) \leq R_{\mathcal{D}_N}(f) + B(N,\mathcal{F})\), et de la forme de cette borne \(B(N,\mathcal{F})\) pour des familles de modèles de capacité différente :
Pour une famille \(\mathcal{F}\) de capacité trop faible (par ex. ici les modèles linéaires) la borne \(B(N,\mathcal{F})\) est basse mais comme le risque empirique (l’erreur d’apprentissage) \(R_{\mathcal{D}_N}(f)\) est élevé(e) nous ne pouvons pas obtenir de garantie intéressante pour le risque espéré \(R(f)\) (pour les performances de généralisation).
Pour une famille \(\mathcal{F}\) de capacité trop élevée (par ex. ici PMC avec \(\alpha = 10^-5\)) le risque empirique \(R_{\mathcal{D}_N}(f)\) est vraisemblablement faible (cette famille a une capacité d’approximation élevée) mais la valeur de la borne \(B(N,\mathcal{F})\) est également élevée. Dans ce cas encore nous ne pouvons pas obtenir de garantie intéressante pour le risque espéré \(R(f)\).
Pour une famille \(\mathcal{F}\) de capacité « adéquate » (par ex. ici PMC avec \(\alpha = 1\)) le risque empirique \(R_{\mathcal{D}_N}(f)\) reste faible et la valeur de la borne \(B(N,\mathcal{F})\) est plutôt basse, l’inégalité \(R(f) \leq R_{\mathcal{D}_N}(f) + B(N,\mathcal{F})\) peut donc nous donner une garantie intéressante pour le risque espéré \(R(f)\) ! Rappelons ici qu’une borne comme celle obtenue dans le théorème précédent est toutefois rarement utile en pratique car elle peut être basse seulement si on dispose de très nombreuses données d’apprentissage (\(N\) très élevé).