L’identification automatique de groupes de données similaires dans un (grand) ensemble de données est une composante importante de la fouille de données. La classification automatique (cluster analysis ou clustering en anglais) cherche à regrouper les données de façon à obtenir des groupes tels que les données sont plus similaires entre elles à l’intérieur d’un même groupe (cluster en anglais) qu’entre groupes. Dans la mesure où les notions de similarité et de groupe peuvent être explicitées de multiples façons, de nombreuses méthodes de classification automatique ont été proposées depuis les années 1930. Des synthèses partielles peuvent être trouvées, par exemple, dans [JMF99] ou [BBS14].
Après une brève typologie des méthodes de classification automatique, nous nous intéresserons à la mise en œuvre d’algorithmes de classification dans un contexte de données massives.
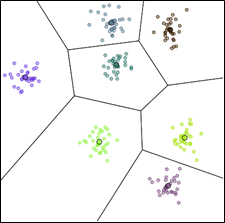
Une première distinction entre méthodes de classification automatique peut être faite suivant leur objectif. La plupart des méthodes visent à obtenir un partitionnement des données, comme dans l’exemple suivant, plus éventuellement de trouver une donnée « représentative » (« prototype ») par groupe :
Fig. 48 Exemple de partitionnement de données bidimensionnelles¶
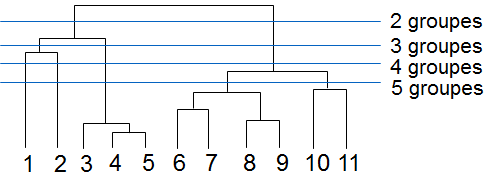
D’autres méthodes cherchent plutôt à obtenir une hiérarchie de regroupements, qui fournit une information plus riche concernant la structure de similarité des données. Noter qu’à partir d’une telle hiérarchie il est facile d’extraire plusieurs partitionnements, à des niveaux de « granularité » différents, comme dans l’exemple suivant :
Fig. 49 Exemple de regroupement hiérarchique qui peut servir à obtenir plusieurs partitionnements¶
Une seconde distinction peut être faite suivant la nature des données à regrouper : données numériques, données catégorielles ou données mixtes. Si la plupart des méthodes visent les données numériques, certaines peuvent traiter directement des données catégorielles. Toutefois, les données catégorielles sont fréquemment transformées en données numériques (par ex. par une analyse factorielle des correspondances multiples) avant d’être traitées par des méthodes de classification adaptées aux données numériques.
Une autre distinction correspond à la représentation des données. De nombreuses méthodes de classification automatique travaillent sur des représentations vectorielles qui, en plus de différentes métriques, permettent de définir des centres de gravité de groupes, des densités de probabilité, des intervalles, des sous-espaces, etc. Certaines méthodes se satisfont, en revanche, d’une simple structure métrique sur l’espace des données ; cela leur confère une grande généralité.
Une distinction importante concerne la nature des groupes recherchés : sont-il mutuellement exclusifs ou non ? Sont-ils « nets » (crisp) ou « flous » (fuzzy) ? Si la plupart des méthodes de classification automatique s’intéressent aux partitionnement exclusifs, certaines permettent d’obtenir des groupes non exclusifs (par ex., dans un mélange gaussien, deux lois peuvent présenter une « intersection » significative). Aussi, des extensions floues existent pour des méthodes de classification automatiques bien connues (par ex. fuzzy c-means pour K-means, voir [JMF99]). Les extensions floues peuvent être plus robustes que les méthodes « nettes » de départ.
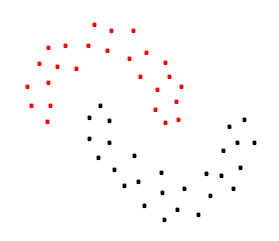
Enfin, une autre distinction importante peut être faite suivant le critère de regroupement. Certaines méthodes cherchent des groupes « compacts » et relativement éloignés entre eux, comme dans cet exemple de partitionnement Fig. 48. D’autres méthodes s’intéressent à des groupes denses (et non nécessairement « compacts ») séparés par des régions moins denses, comme dans l’exemple suivant :
Fig. 50 Exemple de groupes denses séparés par des régions moins denses¶
Il faut noter que ce critère de regroupement n’est pas toujours explicite, pourtant son impact sur les résultats obtenus est majeur.
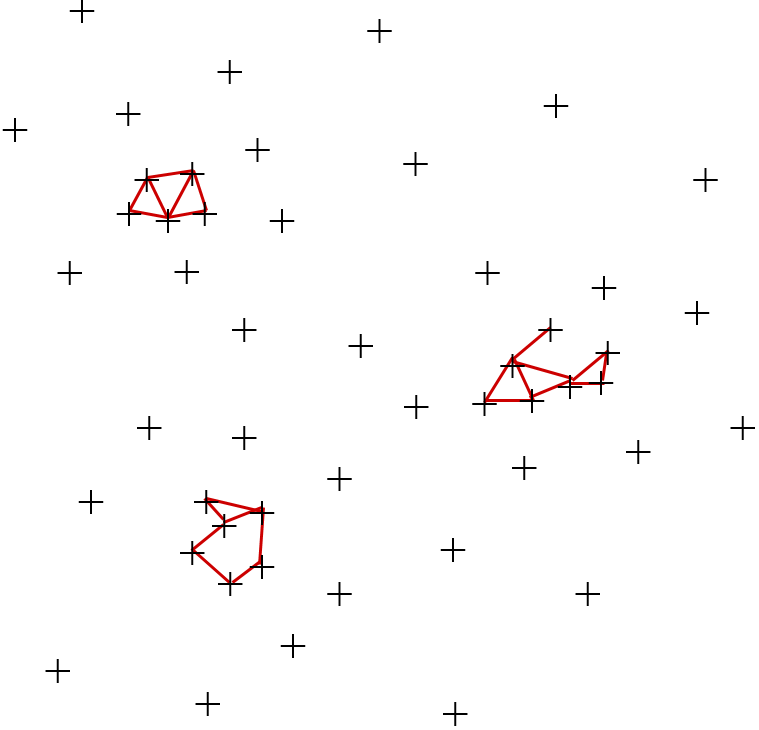
Enfin, revenons sur la distinction entre classification automatique et auto-jointure par similarité. Pour un ensemble de données \(\mathcal{D}\) et un seuil de distance \(\theta\), l’auto-jointure par similarité doit retourner les paires d’éléments de \(\mathcal{D}\) qui sont à une distance inférieure au seuil, c’est à dire \(K_{\theta} = \{(\mathbf{x},\mathbf{y}) | \mathbf{x},\mathbf{y} \in \mathcal{D}, d(\mathbf{x},\mathbf{y}) \leq \theta\}\), voir par exemple la figure suivante :
Fig. 51 Exemple d’auto-jointure par similarité ; les traits correspondent aux liens de forte similarité¶
A partir du graphe résultant il est ensuite possible d’extraire des cliques, des composantes connexes, etc. L’intérêt de cette opération est d’identifier des données hautement similaires, cette forte similarité ayant une signification particulière (par ex. « variantes » d’une même donnée). Les données « isolées », qui n’ont pas de voisin suffisamment proche (au vu de \(\theta\)), sont simplement ignorées dans les résultats.
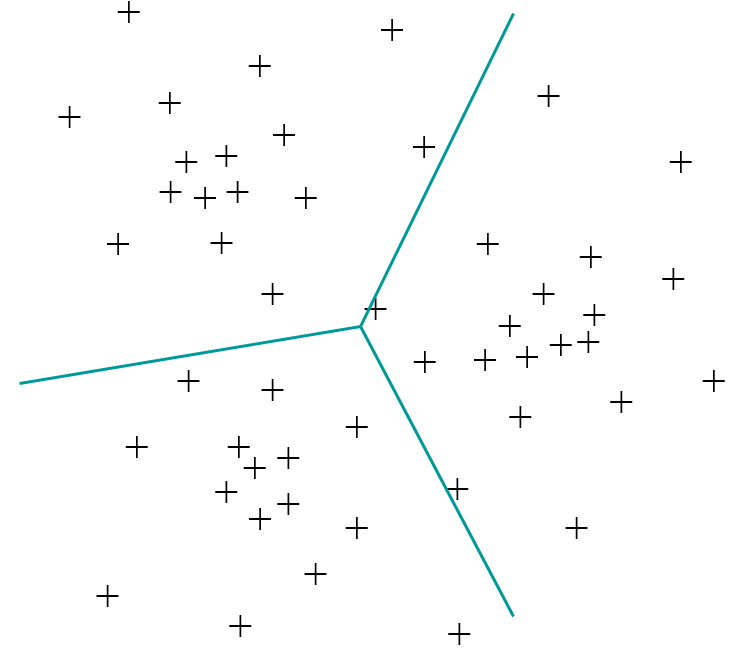
La classification automatique, en revanche, vise un regroupement des données par similarité. En général, chaque donnée appartiendra à un groupe, même si elle n’a pas de voisins proches, comme dans la figure suivante. Il faut toutefois noter que certaines méthodes de classification automatique retournent séparément (hors groupes) des données considérées « isolées » dans le sens « mal expliquées » par tous les groupes trouvés.
Fig. 52 Exemple de classification automatique ; les traits correspondent aux frontières de séparation entre les trois groupes¶
Considérons un ensemble \(\mathcal{E}\) de \(N\) données décrites par \(p\) variables à valeurs dans \(\mathbb{R}\) et \(d\) une distance sur \(\mathbb{R}^p\). Pour regrouper ces données en \(k\) groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\), inconnus a priori, on s’intéresse souvent à un critère qui correspond à la somme des inerties intra-classe des groupes :
avec \(\mathcal{C} = \{\mathbf{m}_j, 1 \leq j \leq k\}\) l’ensemble des centres de gravité des \(k\) groupes. Pour \(\mathcal{E}\) et \(k\) donnés, plus la valeur de \(\phi_\mathcal{E}(\mathcal{C})\) est faible, plus les groupes sont « compacts » autour de leurs centres et donc meilleure est la qualité du partitionnement obtenu.
Trouver le minimum global de la fonction (6) est un problème NP-difficile, mais on dispose d’algorithmes de complexité polynomiale dans le nombre de données \(N\) qui produisent une solution en général sous-optimale.
Un tel algorithme est l’algorithme des centres mobiles décrit ci-dessous :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) ;
Sorties : \(k\) groupes (clusters) disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) et ensemble \(\mathcal{C}\) de leurs centres ;
Affectation de chaque donnée au groupe du centre le plus proche ;
Remplacement des centres par les centres de gravité des groupes ;
fintantque
On peut montrer que la valeur de \(\phi_\mathcal{E}(\mathcal{C})\) diminue lors de chacune des deux étapes du processus itératif (affectation de chaque donnée à un groupe, recalcul des centres). Comme \(\phi_\mathcal{E}(\mathcal{C}) \geq 0\), le processus itératif doit converger. La solution obtenue sera néanmoins un minimum local, dépendant de l’initialisation, souvent de valeur \(\phi_\mathcal{E}(\mathcal{C})\) bien plus élevée que celle correspondant au minimum global (inconnu).
Parfois on appelle k-moyennes (K-means) une version stochastique de la méthode des centres mobiles décrite ci-dessous (les entrées et les sorties sont les mêmes que pour l’algorithme précédent) :
Chaque groupe est initialisé avec son centre comme seul membre du groupe ;
tantque (centres non stabilisés) faire
Choix aléatoire d’une des données ;
Affectation de la donnée au groupe du centre le plus proche ;
Recalcul des centres pour le groupe rejoint par la donnée et le groupe quitté par la donnée ;
fintantque
Dans cette perspective, l’algorithme des centres mobiles est une variante « par lots » (batch) de K-means. Toutefois, K-means est souvent utilisé comme parfait synonyme de l’algorithme des centres mobiles ; c’est ainsi que ce terme sera employé dans la suite du cours.
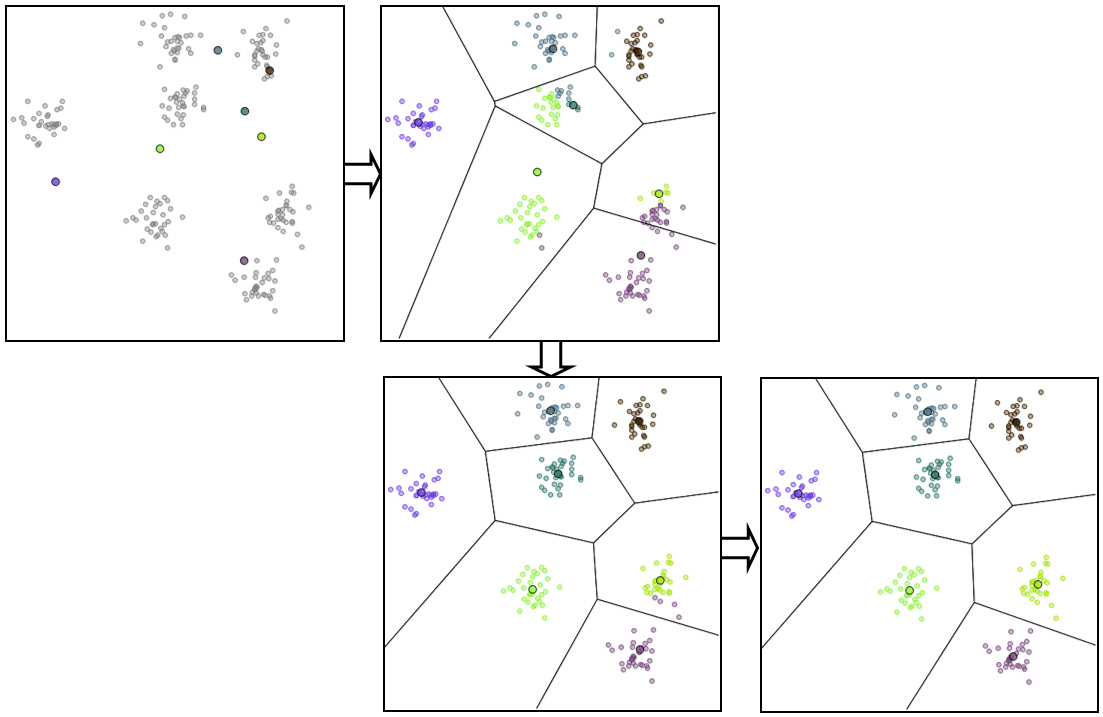
Des étapes successives dans l’application de K-means à des données issues de 7 lois normales bidimensionnelles peuvent être visualisées dans la figure suivante. La distance euclidienne classique est employée et la classification est réalisée avec avec \(k=7\) centres, dont la position initiale (choisie aléatoirement) est indiquée dans la première image.
Fig. 53 Etapes successives dans l’application de K-means¶
Dans cet exemple, la solution obtenue est optimale et est atteinte après seulement 2 itérations.
L’algorithme K-means est facile à exécuter sur une plate-forme distribuée en utilisant le mécanisme MapReduce. Une implémentation simple consiste à assigner aux tâches Map l’affectation des données aux groupes et aux tâches Reduce le recalcul des centres. Cette implémentation est détaillée dans la suite :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) ;
Sorties : \(k\) groupes (clusters) disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) et ensemble \(\mathcal{C}\) de leurs centres ;
L’ensemble \(\mathcal{E}\) de \(N\) données est découpé en fragments distribués aux nœuds de calcul ; un fragment doit tenir dans la mémoire d’un nœud ;
Un nœud initialise les \(k\) centres ;
tantque (centres non stabilisés) faire
Transmettre l’ensemble \(\mathcal{C}\) des centres à tous les nœuds de calcul ;
Chaque tâche Map(t), pour chaque élément \(\mathbf{x}_i\) de son fragment \(t\), trouve le centre le plus proche \(j\) ; ensuite, pour chaque centre \(j\) ainsi trouvé, génère (\(j\), (\(n_{jt}\), \(\tilde{\mathbf{m}}_{jt} = \sum_t \mathbf{x}_i\))), où \(n_{jt}\) est le nombre de données du fragment \(t\) qui ont comme centre le plus proche le centre \(j\) et la somme est faite sur les \(\mathbf{x}_i\) du fragment \(t\) plus proches du centre \(j\) ;
Les paires (\(j\), (\(n_{jt}\), \(\tilde{\mathbf{m}}_{jt}\))) sont groupées par \(j\) pour les tâches Reduce ;
Chaque tâche Reduce reçoit toutes les paires correspondant à une valeur de \(j\), calcule \(\mathbf{m}_j = \frac{\sum_t \tilde{\mathbf{m}}_{jt}}{\sum_t n_{jt}}\) et stocke le \(\mathbf{m}_j\) résultant ;
fintantque
Une variante stochastique peut être facilement obtenue en utilisant des échantillons des données (plutôt que la totalité des données de chaque fragment) dans les tâches Map.
L’initialisation des centres est une étape critique de K-means. La figure suivante illustre les résultats obtenus avec K-means à partir de trois autres initialisations aléatoires des centres :
Fig. 54 Résultats différents avec initialisations aléatoires différentes de K-means¶
On constate que ces trois solutions sont sous-optimales : pour chacune, un des groupes « naturels » (on peut en juger visuellement car les données sont bidimensionnelles et peu nombreuses, ce ne sera pas le cas en général) est divisé en deux, et deux des groupes « naturels » sont regroupés. Les valeurs de \(\phi_\mathcal{E}(\mathcal{C})\) sont plus élevées pour ces trois résultats que pour le résultat optimal illustré plus haut. Exécuter K-means plusieurs fois, à partir d’initialisations aléatoires différentes, ne garantit pas de trouver la solution optimale (ou, dans le cas général, une solution proche de la solution optimale).
Une bonne initialisation de l’algorithme K-means doit permettre d’obtenir une solution de meilleure qualité mais aussi une convergence plus rapide (c’est à dire avec moins d’itérations) vers cette solution.
Parmi les nombreux algorithmes d’initialisation de K-means nous considérerons ici K-means++ proposé dans [AV07]. L’idée de départ est assez simple : choisir les centres l’un après l’autre, suivant une loi non uniforme qui évolue après le choix de chaque centre et qui privilégie les candidats éloignés des centres déjà sélectionnés. L’algorithme d`initialisationK-means++ est :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) et nombre souhaité de centres \(k\) ;
Sorties : \(\mathcal{C} = \{\mathbf{c}_j, 1 \leq j \leq k\}\), à utiliser après comme centres de départ dans K-means ;
\(\mathcal{C} \leftarrow\) un \(\mathbf{x}\) de \(\mathcal{E}\) choisi au hasard ;
tantque (\(\|\mathcal{C}\| \lt k\)) faire
Sélectionner \(\mathbf{x} \in \mathcal{E}\) avec la probabilité \(\frac{d^2(\mathbf{x},\mathcal{C})}{\phi_\mathcal{E}(\mathcal{C})}\) ;
Dans cet algorithme, nous avons noté \(d^2(\mathbf{x},\mathcal{C}) = \min_{j=1,\ldots,\|\mathcal{C}\|} d^2(\mathbf{x},\mathbf{c}_j)\), où \(\|\mathcal{C}\|\) est le nombre de centres déjà choisis. La fonction (6) peut donc être écrite \(\phi_\mathcal{E}(\mathcal{C}) = \sum_{\mathbf{x} \in \mathcal{E}} d^2(\mathbf{x},\mathcal{C})\).
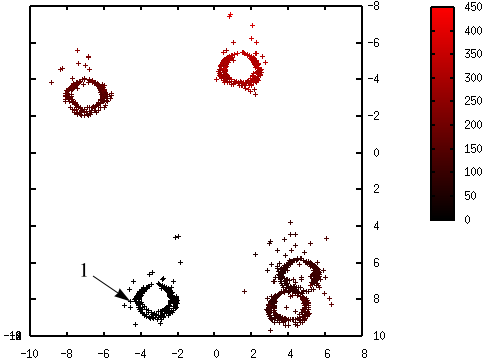
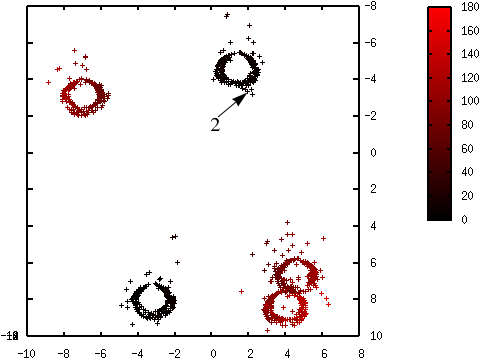
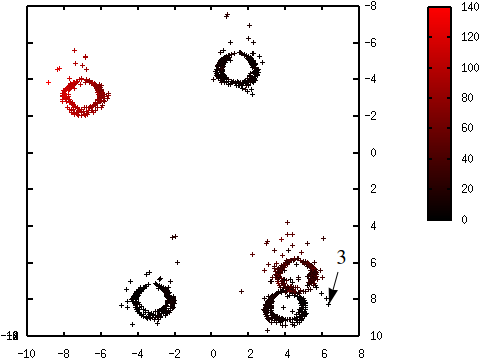
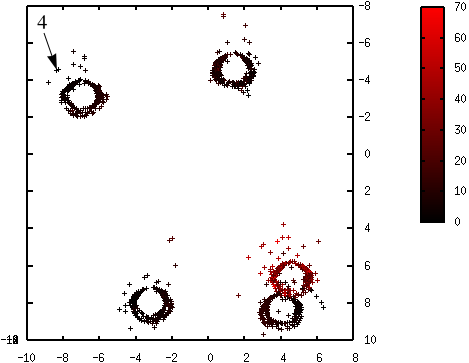
Les figures suivantes illustrent l’évolution des probabilités lors du déroulement de l’algorithme K-means++. La probabilité de sélection d’une donnée dans l’étape suivante (proportionnelle à \(d^2(\mathbf{x},\mathcal{C})\)) est d’autant plus élevée que la couleur est proche du rouge. La première coordonnée correspond à l’axe horizontal, la seconde à l’axe vertical.
Fig. 55 Probabilités après la sélection du premier centre (en bas à gauche) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{c}-4,6\\8,0\end{array}\right)\right\}\)¶
Fig. 56 Probabilités après la sélection du deuxième centre (en haut à droite) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{cc}-4,6 & 2,15\\8,0 & -3,45\end{array}\right)\right\}\)¶
Fig. 57 Probabilités après la sélection du troisième centre (en bas à droite) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{ccc}-4,6 & 2,15 & 6,32\\8,0 & -3,45 & 8,22\end{array}\right)\right\}\)¶
Fig. 58 Probabilités après la sélection du quatrième centre (en haut à gauche) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{cccc}-4,6 & 2,15 & 6,32 & -8,37\\8,0 & -3,45 & 8,22 & -4,54\end{array}\right)\right\}\)¶
La sélection séquentielle des centres opérée par K-means++ permet de bien répartir les centres dans les données. Cela diminue le risque d’avoir, d’une part, des groupes de données sans aucun centre à proximité au démarrage de K-means et, d’autre part, plusieurs centres très proches entre eux. Ce sont ces insuffisances d’une initialisation aléatoire uniforme qui avaient provoqué la convergence à des solutions sous-optimales dans les cas simples illustrés plus tôt Fig. 54.
Malheureusement, K-means++ n’est pas directement parallélisable ; dans le cas de données massives, le nombre \(N\) de données et, implicitement, le nombre \(k\) de centres nécessaires peuvent être très élevés. Employer K-means++ pour générer de l’ordre de \(10^5\) centres ou plus ralentirait significativement l’opération de classification automatique. Comment conserver les bonnes propriétés de l’initialisation par K-means++ tout en permettant de l’accélérer grâce à une exécution distribuée ?
L’algorithme K-means|| a été proposé dans [BMV12] comme variante parallélisable de K-means++. Son principe est de choisir bien plus qu’un seul centre à chaque itération d’initialisation, suivant également une loi non uniforme. On obtient beaucoup plus de centres que nécessaire (taux de sur-échantillonnage \(l \in \Omega(k)\)), ensuite on applique K-means pour regrouper ces centres en \(k\) groupes dont les centres seront employés dans l’application de K-means à la totalité des données.
K-means|| est l’algorithme suivant :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\), nombre souhaité de centres \(k\) et taux de sur-échantillonnage \(l \sim \Omega(k)\) (\(l\) augmente au moins aussi vite que \(k\));
Sorties : \(\mathcal{C} = \{\mathbf{c}_j, 1 \leq j \leq k\}\), à utiliser après comme centres de départ dans K-means ;
\(\mathcal{C} \leftarrow\) un \(\mathbf{x}\) de \(\mathcal{E}\) choisi au hasard ;
\(\mathcal{C}' \leftarrow\) sélectionner chaque \(\mathbf{x} \in \mathcal{E}\) indépendamment avec la probabilité \(\frac{l \cdot d^2(\mathbf{x},\mathcal{C})}{\psi}\) ;
\(w_m \leftarrow\) nombre de données de \(\mathcal{E}\) plus proches de \(\mathbf{m}_j\) que de tout autre point de \(\mathcal{C}\) ;
finpour
Classification en \(k\) groupes des données \(\mathcal{C}\)pondérées par leurs poids\(w_m\). Les centres de ces groupes, en nombre de \(k\), formeront l’ensemble final de centres \(\mathcal{C} = \{\mathbf{c}_j, 1 \leq j \leq k\}\) à utiliser ensuite comme initialisation de K-means.
L’algorithme démarre par le choix aléatoire du premier centre. Ensuite, lors de chaque itération de (3.1)-(3.2), plusieurs données sont sélectionnées comme centres potentiels, suivant une loi qui rend plus probable le choix de candidats éloignés des centres déjà présents, et ensuite ajoutées à l’ensemble des candidats \(\mathcal{C}\). Lors de l’étape (3.1), \(\sum_{\mathbf{x} \in \mathcal{E}} \frac{l \cdot d^2(\mathbf{x},\mathcal{C})}{\psi} = l\), donc à chaque itération env. \(l\) nouvelles données sont choisies.
Dans l’étape (4), les candidats centres de \(\mathcal{C}\) sont pondérés. Le poids de chaque candidat est le nombre de données de l’ensemble global \(\mathcal{E}\) qui sont plus proches de ce candidat que de tout autre candidat. Ce poids est donc une mesure de l’importance du candidat. Si de nombreuses données de \(\mathcal{E}\) sont « représentées » par un même candidat, il faudrait avoir au moins un centre à proximité de ce candidat. En revanche, si un candidat « représente » peu de données alors soit il est dans une région isolée dans l’espace qui ne « mérite » pas d’avoir un centre à proximité, soit il est trop proche d’autres candidats et devrait donc partager un même centre avec eux.
L’étape finale (5) consiste à faire une classification automatique des candidats de \(\mathcal{C}\) en \(k\) groupes. Les données de \(\mathcal{C}\) interviennent pondérées : pour chaque itération de l’algorithme de classification, les données affectées au groupe sont pondérées lors du calcul du centre de gravité du groupe. L’étape (5) traite un nombre relativement réduit de données, \(O(l \cdot \log \psi)\), elle peut donc se dérouler sur un seul nœud de calcul et employer un algorithme séquentiel comme K-means++ pour choisir les centres.
Comme K-means++, K-means|| donne des garanties de qualité de la solution obtenue (voir [BMV12]) : \(E[\phi_\mathcal{E}(\mathcal{C})] \leq O(\log k) \cdot \phi_\mathcal{E}(\mathcal{C}^*)\), \(E\) étant l’espérance et \(\mathcal{C}^*\) la solution optimale. Noter que les groupes sont définis sans équivoque par l’ensemble des centres, le partitionnement optimal est celui défini par l’ensemble optimal de centres \(\mathcal{C}^*\).
D’après [BMV12], cinq itérations dans l’étape (3) suffisent en général pour atteindre une très bonne solution, il n’est pas indispensable d’effectuer \(O(\log \psi)\) itérations.
L`implémentation MapReduce de l’algorithme d’initialisation K-means|| est facile. Chaque itération de (3) est exécutée de façon distribuée ; les tâches Map effectuent les opérations de (3.1), c’est à dire le calcul des \(d^2(\mathbf{x},\mathcal{C})\) et les tirages, ainsi qu’une partie du calcul de \(\psi\) sur les données du fragment local ; les tâches Reduce finalisent le calcul de \(\psi\) (comme dans l’implémentation MapReduce de K-means). Comme pour l’implémentation de K-means, après sa mise à jour dans l’étape (3.2), l’ensemble \(\mathcal{C}\) des candidats centres est transmis à tous les nœuds de calcul pour l’étape (3.1).
Dans l’étape (4), le calcul des poids \(w_m\) est réalisé comme le calcul des \(n_j\) dans l’implémentation MapReduce de K-means : les tâches Map trouvent l’élément de \(\mathcal{C}\) le plus proche de chaque donnée de leur fragment, puis calculent des valeurs partielles des \(w_m\) sur leur fragment. Les cumuls entre fragments sont ensuite réalisés par les tâches Reduce.
La classification des données (candidats centres) de \(\mathcal{C}\) en \(k\) groupes dans l’étape (5) peut se dérouler sur un seul nœud de calcul et employer un algorithme séquentiel comme K-means++ pour choisir les centres. Les centres résultants, \(\mathcal{C} = \{\mathbf{c}_j, 1 \leq j \leq k\}\), sont conservés et transmis comme centres initiaux à l’algorithme K-means.
Classification descendante hiérarchique avec Bisecting k-means¶
Parmi les méthodes qui visent à obtenir une hiérarchie de regroupements, nous examinons brièvement dans la suite Bisecting k-means qui est une méthode de classification descendante hiérarchique. Rappelons que, par rapport à un simple partitionnement des données, une hiérarchie de regroupements fournit une information plus riche concernant la structure de similarité des données. A partir de la hiérarchie de groupes résultante il est possible d’observer l’ordre des agrégations de groupes et d’examiner les rapports de similarités entre groupes. Une hiérarchie de groupes peut également permettre d’obtenir plusieurs partitionnements à des niveaux de granularité différents.
Afin d’obtenir une classification hiérarchique nous pouvons employer une approche ascendante ou une approche descendante. La classification ascendante procède par agrégations successives des groupes les plus proches. Différentes mesures de distance entre groupes peuvent être obtenues à partir des distances entre données individuelles. Lorsque le point de départ de la classification ascendante hiérarchique (CAH) est constitué par l’ensemble des données individuelles (en nombre de \(N\)), la CAH présente une complexité algorithmique de \(O(N^2)\), supérieure à celle de K-means. Pour cette raison, elle est plutôt appliquée sur les centres des groupes issus d’une classification par K-means avec un nombre très élevé de groupes (groupes de granularité fine). Enfin, la CAH n’est pas efficacement parallélisable, raison pour laquelle il n’y a pas d’implémentation Spark reconnue.
La classification descendante procède plutôt par découpages récursifs des données et s’arrête en général bien avant d’atteindre le niveau des données individuelles. Si la méthode de découpage employée est bien choisie, la classification descendante hiérarchique (CDH) peut garder une complexité réduite et être efficacement parallélisable. Une solution simple consiste à se servir de K-means pour séparer des groupes obtenus aux itérations précédentes en sous-groupes. Cette solution est employée dans la méthode bisecting k-means qui est présentée dans la suite.
Bisecting k-means procède par le découpage, à chaque itération, du groupe de données le moins compact en deux (sous-)groupes obtenus par application de K-means avec \(k = 2\). L’algorithme est le suivant :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) et nombre souhaité de groupes \(k\) ;
Initialization: liste avec un seul groupe qui regroupe toutes les données ;
tantque (nombre groupes \(\lt k\)) faire
Retirer de la liste le groupe qui a la plus forte inertie intra-classe ;
pour (\(i\) de 1 à nb_essais) faire
Appliquer K-means avec \(k = 2\) pour diviser ce groupe en 2 sous-groupes ;
finpour
Ajouter à la liste des groupes les 2 sous-groupes avec la plus faible somme des inerties intra-classe ;
fintantque
On observe que la somme des inerties intra-groupe diminue à chaque itération, le groupe le moins compact (qui a la plus forte inertie intra-classe) étant divisé en deux sous-groupes. Aussi, le découpage fait à chaque itération est éventuellement affiné dans les itérations ultérieures mais n’est pas remis en cause, donc des données qui sont séparées du « bon » groupe à une itération ne peuvent pas retourner dans ce groupe après.
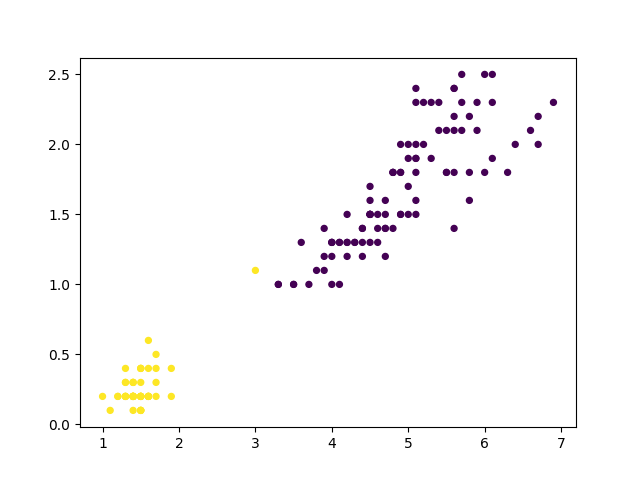
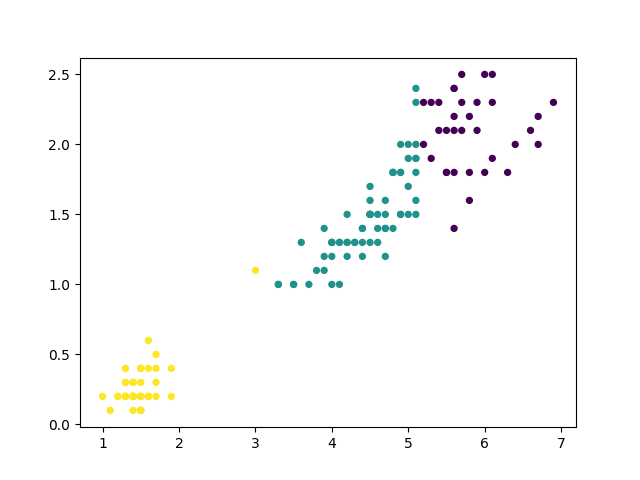
Les deux figures suivantes illustrent les résultats de deux itérations successives sur un eptit ensemble de données (les sépales des Iris).
Fig. 59 Après la première itération de bisecting k-means sont obtenus deux groupes. On observe qu’un des points (en jaune) n’est pas affecté au groupe le plus approprié. C’est une illustration du fait que l’application de K-means avec \(k=2\) peut produire des découpages trop grossiers, qui sont affinés mais pas remis en cause lors des itérations ultérieures.¶
Fig. 60 Après la seconde itération de bisecting k-means sont obtenus trois groupes. C’est bien le groupe qui présentait l’inertie intra-classe la plus élevée qui a été découpé en deux.¶
Par rapport à K-means, bisecting k-means présente une meilleure stabilité car à chaque itération plusieurs essais sont réalisés pour découper le groupe qui présente l’inertie intra-classe la plus élevée et les groupes conservés sont les deux sous-groupes dont la somme des inerties intra-classe est la plus faible. Par ailleurs, le coût de bisecting k-means est plus faible que celui de k-means car seule la première itération travaille sur toutes les données (et pour trouver seulement deux groupes), chacune des itérations suivantes se limite à la partie des données correspondant au groupe le moins compact.
En revanche, les résultats obtenus avec K-means peuvent être mieux ajustés aux groupes présents dans les données que les résultats obtenus avec bisecting k-means. En effet, l’application de K-means avec \(k=2\) lors des premières itérations de bisecting k-means peut produire des découpages trop grossiers qui coupent des groupes présents dans les données, or ces découpages ne sont pas remis en cause lors des itérations ultérieures de bisecting k-means.
Spark propose des implémentations de plusieurs algorithmes de classification automatique :
K-means, avec une initialisation par K-means|| ([BMV12]) que nous avons étudié plus haut.
Estimation de mélanges gaussiens en utilisant l’algorithme espérance-maximisation (Expectation-Maximization, EM). Le mélange gaussien résultant est une estimation de densités qui peut également servir à la classification automatique : chaque donnée sera « affectée » au groupe défini par la composante du mélange qui « explique » le mieux cette donnée. EM est un algorithme itératif, chaque itération comporte une étape de calcul de l’espérance de la vraisemblance et une étape de calcul des paramètres (paramètres des lois normales du mélange et coefficients de mélange) qui maximisent cette vraisemblance. Les équations de mise à jour sont facilement parallélisables.
Bisecting K-means, méthode de classification descendante hiérarchique qui à chaque itération partitionne en deux sous-groupes le groupe d’inertie intra-classe la plus élevée. Nous l’avons étudié plus haut.
Power Iteration Clustering (PIC, voir [LC10]) est une simplification de la classification spectrale qui travaille sur la matrice des similarités entre données.
Pour Latent Dirichlet Allocation (LDA, voir [BNJ03]), l’objectif initial était d’identifier des « thèmes » (topics) dans un ensemble de documents textuels et ensuite d”« expliquer » chaque document par un ou plusieurs de ces thèmes. La méthode a toutefois une applicabilité plus large.
Arthur, D. and S. Vassilvitskii. K-means++: The advantages of careful seeding. In Proceedings of the 18th Annual ACM-SIAM Symposium on Discrete Algorithms, SODA’07, pages 1027–1035, Philadelphia, PA, USA, 2007. Society for Industrial and Applied Mathematics.
Bouveyron, C., C. Brunet-Saumard. Model-based clustering of high-dimensional data: A review, Computational Statistics and Data Analysis, Volume 71, March 2014, Pages 52-78, ISSN 0167-9473, http://dx.doi.org/10.1016/j.csda.2012.12.008.
Jain, A. K., M. N. Murty, and P. J. Flynn. Data clustering: a review. ACM Comput. Surv. 31, 3 (September 1999), 264-323. DOI=http://dx.doi.org/10.1145/331499.331504.
Lin, F. and W. W. Cohen. Power iteration clustering. In J. Fürnkranz and T. Joachims, editors, International Conference on Machine Learning, pages 655–662. Omnipress, 2010.