Un modèle décisionnel doit permettre de prédire, pour chaque nouvelle donnée (ou observation), la valeur d’une variable expliquée (ou « dépendante », ou « de sortie ») à partir des valeurs prises par les autres variables (« explicatives », ou « d’entrée ») pour cette donnée. Dans un problème de discrimination entre deux ou plusieurs classes, la variable expliquée est une variable nominale, dont chaque modalité correspond à une des classes possibles. Dans un problème de régression, la variable expliquée est une variable quantitative. Dans un problème de prédiction structurée, la variable expliquée prend des valeurs dans un domaine de données structurées (par ex. de graphes).
Un modèle décisionnel peut être construit analytiquement, à partir d’une parfaite compréhension du phénomène observé, ou par des moyens statistiques, sur la base d’un ensemble d’observations disponibles. Parfois on a une compréhension partielle du phénomène observé, qui permettra de définir des a priori utiles pour l’identification statistique du modèle. L’apprentissage supervisé vise à construire un modèle décisionnel à partir de données pour lesquelles une information de « supervision » est disponible. Cette information correspond en général, pour chaque donnée, à la valeur prise par la variable expliquée.
Avant d’aller plus loin, rappelons que la prise de décision peut aussi être faite à partir d’un ensemble d’observations disponibles sans construction préalable d’un modèle. Pour décider de la valeur de la variable expliquée pour une nouvelle donnée il est possible d’utiliser, par exemple, directement les k plus proches observations pour lesquelles la valeur de la variable expliquée est connue.
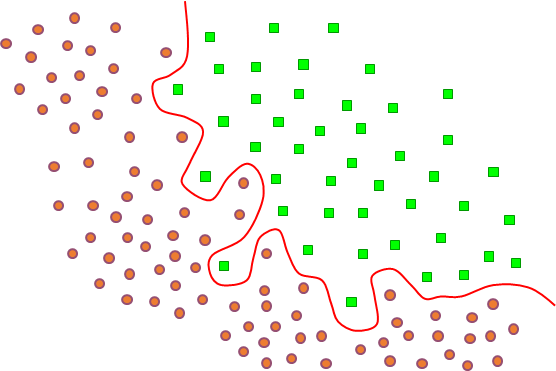
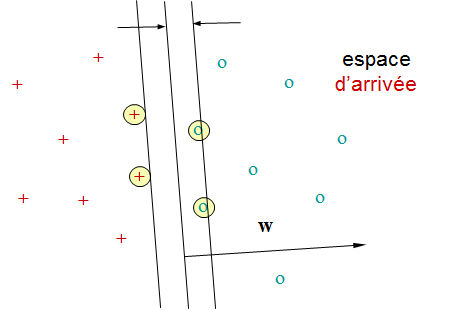
La figure suivante illustre un problème de discrimination entre deux classes dans \(\mathbb{R}^2\). La variable expliquée possède une modalité pour chaque classe. Les points clairs sont les données ayant permis de construire de modèle et le trait noir est la frontière de discrimination à laquelle peut se réduire ici le modèle décisionnel. Toute nouvelle donnée se situera d’un côté ou de l’autre de cette frontière et sera ainsi affectée par le modèle à une des classes. Ce modèle peut éventuellement être complété par des critères de rejet (refus d’affectation), par ex. on peut refuser de classer les données trop proches de la frontière (rejet d’ambiguïté) ou trop éloignées des données ayant permis de construire de modèle (rejet de non représentativité).
Fig. 66 Exemple de discrimination entre deux classes dans \(\mathbb{R}^2\). Les points clairs sont les données d’apprentissage et le trait noir la frontière de discrimination obtenue.¶
L’apprentissage supervisé n’est pas la seule approche de construction de modèles à partir de données. Nous pouvons mentionner, entre autres :
L’apprentissage non supervisé. Dans ce cas, aucune information de supervision n’est disponible, par ex. on ne connaît la classe d’appartenance pour aucune des données disponibles. Souvent, on ne sait même pas quelles sont les classes pertinentes pour caractériser les données. La classification automatique fait partie de cette famille. Le modèle obtenu par apprentissage non supervisé est en général employé comme modèle descriptif et aide à mieux définir un problème décisionnel qui est traité ultérieurement (par ex., à mettre en évidence des groupes ou clusters qui ont un rapport étroit avec une ou plusieurs classes dont la détection serait utile).
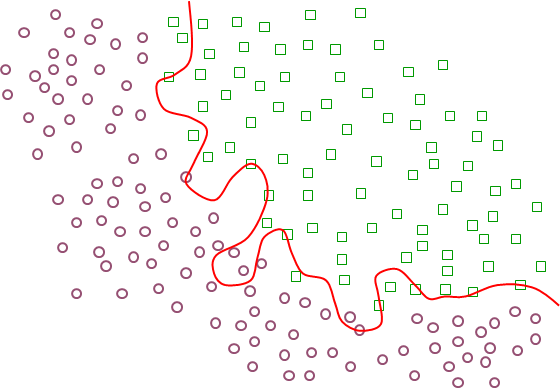
L’apprentissage semi-supervisé (voir par ex. [CSZ06]). Dans ce cas, l’information de supervision est disponible sous une forme partielle, par ex. pour une (faible) partie des données d’apprentissage. Par ailleurs, la connaissance a priori du problème traité permet éventuellement de valider des hypothèses sur le lien entre la distribution des données et la variable expliquée (par ex., la séparation entre classes correspond à des régions de faible densité, comme dans la figure précédente). Ces hypothèses permettent de se servir des données qui n’ont pas d’information de supervision afin d’améliorer la modélisation décisionnelle par rapport à l’utilisation exclusive des données pour lesquelles la supervision est présente.
Dans la suite de ce chapitre nous nous intéresserons d’abord à la formulation d’un problème d’apprentissage supervisé et à l’évaluation des performances de généralisation du modèle décisionnel ainsi obtenu. Seront étudiées également les questions de passage à l’échelle et leurs implications pour l’apprentissage supervisé. Nous examinerons ensuite une famille très populaire d’outils de modélisation décisionnelle, les machines à vecteurs de support. Pour trouver le modèle à partir des données d’apprentissage il est en général nécessaire de choisir les valeurs d’un ou plusieurs hyperparamètres (qui permettent de mieux définir la famille de modèles candidats) et de procéder à une optimisation pour déterminer les paramètres du modèle. Les hyperparamètres ont un impact significatif sur les résultats, nous regarderons comment faire la recherche des meilleures valeurs pour ces hyperparamètres. Enfin, nous passerons brièvement en revue les outils proposés par la plate-forme Spark pour l’apprentissage supervisé. L’utilisation de machines à vecteurs de support dans Spark sera abordée dans la séance de travaux pratiques.
Considérons un espace d’entrée \(\mathcal{X}\), dans lequel prennent des valeurs les variables explicatives, et un espace de sortie \(\mathcal{Y}\) dans lequel prend des valeurs la variable expliquée (ou variable « dépendante »).
Les variables explicatives numériques sont en général représentées par des variables réélles et l’espace d’entrée est \(\mathbb{R}^p\) (\(p\) étant le nombre de variables explicatives numériques). Les variables explicatives nominales peuvent être représentées par des variables numériques, à condition de ne pas « travestir » leur nature. Par exemple, entre les modalités d’une variable nominale « catégorie socio-professionnelle » il n’y a pas de relation d’ordre ni de similarité directement quantifiable. Représenter ces modalités par des valeurs différentes d’une même variable numérique introduirait à la fois une relation d’ordre et des similarités quantifiables, étrangères à la nature de la variable de départ et avec un impact négatif fort sur la modélisation. Une variable nominale doit plutôt être représentée à travers un codage disjonctif, avec une variable binaire par modalité (1 pour la modalité présente, 0 pour les autres). Il est également possible d’appliquer en amont une analyse des correspondances multiples aux données décrites par des variables nominales afin de les représenter par les variables quantitatives qui sont les facteurs issus de l’analyse (en nombre en général bien plus faible que les variables nominales de départ).
Lorsque les valeurs des variables explicatives sont des données structurées (par ex. des arbres d’analyse grammaticale ou des graphes de structure de protéines), leur représentation par des ensembles de variables quantitatives est plus problématique. Des outils de modélisation comme les machines à vecteurs de support peuvent employer des fonctions-noyau spécifiques à de tels types de données, sans passer par une représentation vectorielle intermédiaire.
Pour un problème de discrimination entre deux classes, l’espace de sortie est souvent \(\{-1;1\}\). Lorsque le nombre de classes est supérieur à deux, le problème est le plus souvent décomposé en un ensemble de problèmes à deux classes, comme nous le verrons plus loin. Pour un problème de régression, l’espace de sortie est en général \(\mathbb{R}\).
Pour la modélisation on dispose des données \(\mathcal{D}_N = \{(\mathbf{x}_i, y_i)\}_{1 \leq i \leq N}\), l’information de supervision correspondant aux valeurs \(\{y_i\}_{1 \leq i \leq N}\). On considère en général que ces données sont des réalisations des variables aléatoires \(\mathcal{D}_N = \{(X_i, Y_i)\}_{1 \leq i \leq N}\), où \((X_i,Y_i) \in \mathcal{X} \times \mathcal{Y}\), \((X_i,Y_i)\) respectent la loi (inconnue) \(P\) pour tout \(i\) et \((X_i,Y_i)\) sont indépendantes de \((X_j,Y_j)\) pour \(i \neq j\).
L’objectif de l’apprentissage supervisé est d’obtenir le modèle décisionnel qui présente la meilleure généralisation, à partir de données pour lesquelles l’information de supervision est disponible. Cela revient en général à trouver, dans une famille de fonctions \(\mathcal{F}\), une fonction \(f : \mathcal{X} \rightarrow \mathcal{Y}\) qui « prédit » \(y\) à partir de \(\mathbf{x}\) en minimisant le risque espéré (ou risque théorique, ou erreur de généralisation) :
Dans cette expression, \(E_P\) est l’espérance par rapport à la loi inconnue \(P\) et \(L\) est la fonction de « perte » ou d’erreur.
Les fonctions de « perte » (ou d’erreur) \(L\) les plus fréquemment employées sont :
La perte quadratique pour la régression : \(L_Q(\mathbf{x},y,f) = [f(\mathbf{x})-y]^2\), \(\mathbf{x}\) étant l’observation d’entrée, \(f(\mathbf{x})\) la prédiction du modèle et \(y\) l’information de supervision associée à cette observation. L’erreur est donc le carré de la différence entre la valeur prédite par le modèle et la valeur observée.
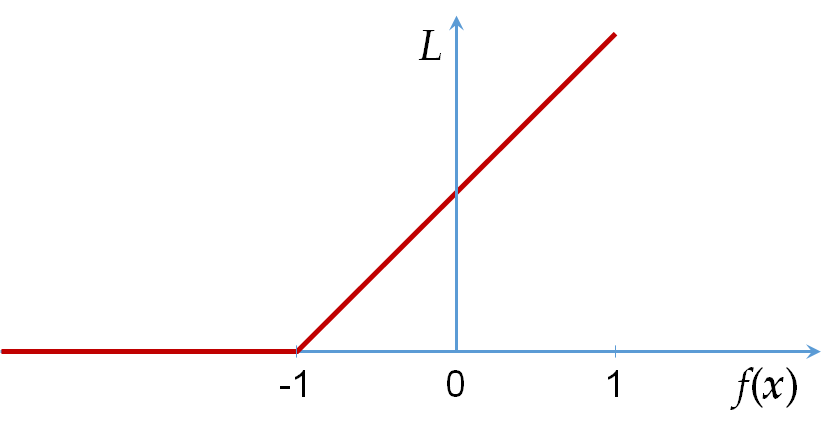
La perte 0-1 pour la discrimination entre 2 classes : \(L_{0-1}(\mathbf{x},y,f) = \mathbf{1}_{f(\mathbf{x}) \neq y}\), \(y \in \{-1;1\}\). L’erreur est nulle si la classe prédite est la même que la classe observée, et égale à 1 sinon.
Hinge loss pour la discrimination entre 2 classes en maximisant la marge (nous y reviendrons dans la section qui traite les machines à vecteurs de support) : \(L_H(\mathbf{x},y,f) = \max\{0, 1 - y f(\mathbf{x})\}\), \(y \in \{-1;1\}\). Contrairement à la perte 0-1, hinge loss est continue et différentiable partout sauf dans \(y f(\mathbf{x}) = 1\). Des variantes différentiables partout (« lissées ») existent également.
Pour obtenir un modèle décisionnel à partir des données de \(\mathcal{D}_N\), il est nécessaire de choisir une famille paramétrique \(\mathcal{F}(\mathbf{w})\) de modèles (par ex. les modèles linéaires) et ensuite d’appliquer un algorithme d’optimisation pour déterminer le (vecteur de) paramètre(s) optimal \(\mathbf{w}^*\) qui définit le modèle dans la famille \(\mathcal{F}(\mathbf{w})\).
L’objectif est de trouver le modèle qui présente la meilleure généralisation. Comme le risque espéré \(R(f)\) d’un modèle \(f\) ne peut pas être directement évalué car \(P\) est inconnue, il n’est pas envisageable de déterminer le modèle en minimisant directement son risque espéré. En revanche, il est possible de mesurer le risque empirique de \(f\), c’est à dire l’erreur de \(f\) sur les données de \(\mathcal{D}_N\) :
Le modèle \(f \in \mathcal{F}\) étant défini par un paramètre \(\mathbf{w}\), le risque empirique \(R_{\mathcal{D}_N}(f)\) est également directement exprimable comme une fonction de ce paramètre et il est alors envisageable de chercher le paramètre optimal \(\mathbf{w}^*\) qui minimise le risque empirique :
Dans quelle mesure cette minimisation du risque empirique permet d’obtenir un modèle qui a un risque théorique minimal ? Il faut examiner deux aspects différents :
La cohérence (consistency) de la minimisation du risque empirique (MRE) : quand le nombre \(N\) de données d’apprentissage tend vers l’infini, le risque empirique (l’erreur d’apprentissage) converge-t-il vers le risque espéré (l’erreur de généralisation) ? Différents résultats existent concernant cette question (voir par ex. [BBL99]) ; on peut ainsi montrer que la MRE est cohérente si et seulement si la VC-dimension (la dimension de Vapnik-Chervonenkis) de la famille \(\mathcal{F}\) (dans laquelle on cherche \(f\)) est finie.
Si le nombre \(N\) de données d’apprentissage est fini, est-ce possible de borner l’écart entre le risque espéré et le risque empirique pour un modèle \(f\) ? On peut montrer (voir par ex. [Vap98], [BBL99]) que de telles bornes existent et dépendent à la fois de la « capacité » de \(\mathcal{F}\) (par ex. de sa VC-dimension) et du nombre \(N\) de données d’apprentissage : \(R(f) \leq R_{\mathcal{D}_N}(f) + B(N,\mathcal{F})\). La borne \(B(N,\mathcal{F})\) diminue (est plus stricte) lorsque \(\mathcal{F}\) est de « capacité » plus faible et lorsque \(N\) augmente, mais reste en général trop élevée pour permettre de borner \(R(f)\) de façon utile.
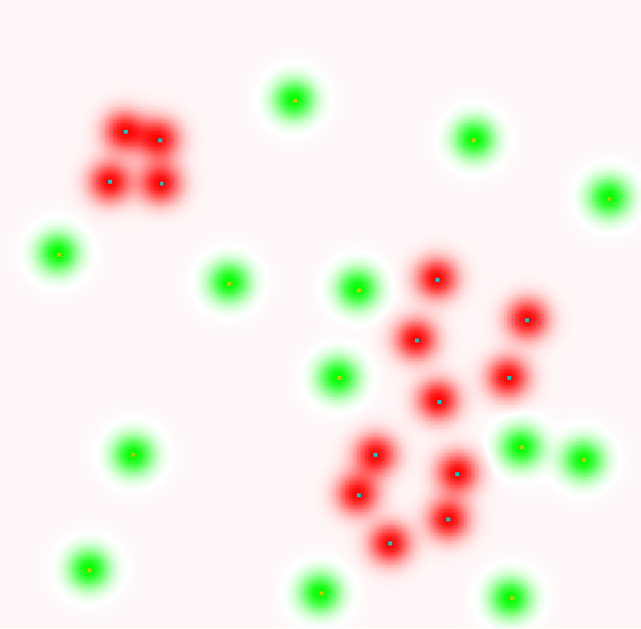
Ces résultats sont intéressants d’un point de vue théorique mais leur utilité pratique est limitée. Avec un ensemble de données d’apprentissage \(\mathcal{D}_N\) fini, la minimisation du risque empirique peut avoir comme résultat un modèle qui généralise mal (sur-apprentissage ou apprentissage « par cœur ») même pour une famille \(\mathcal{F}\) de capacité finie et relativement faible. Les deux figures suivantes illustrent un tel cas où le modèle (la frontière de discrimination entre deux classes) issue de la MRE a un risque empirique nul mais une erreur de généralisation (sur des données non employées pour l’apprentissage) comparativement élevée.
Fig. 68 Erreur comparativement élevée sur des données de test (non utilisées pour l’apprentissage)¶
Une solution fréquemment adoptée est la minimisation du risque empirique régularisé (MRER), qui cherche à réduire le risque empirique tout en pénalisant la « complexité » du modèle :
Si la minimisation du seul risque empirique mène à une augmentation de la complexité du modèle, le terme de régularisation \(\textrm{Reg}(f(\mathbf{w}))\) doit pénaliser plus ce modèle. Le résultat est un compromis entre la réduction de l’erreur d’apprentissage et l’augmentation de la complexité du modèle. Le terme de régularisation cherche, par exemple, à privilégier les modèles « lisses » et son expression exacte peut dépendre de la nature de la famille \(\mathcal{F}\).
Une approche alternative est la minimisation du risque structurel (MRS) : on considère une séquence \(\mathcal{F}_d\), \(d \in \mathbb{N}\) de modèles de « capacité » \(d\) croissante, dans chaque famille on minimise le risque empirique, ensuite on pénalise la capacité de la famille. Sont ainsi obtenus plusieurs modèles dans des familles de capacité croissante, le modèle retenu est celui qui présente le meilleur compromis entre risque empirique et capacité :
Sans garanties a priori sur le risque espéré (l’erreur de généralisation) d’un modèle obtenu par MRE ou MRER, il est important de pouvoir estimer ce risque afin de savoir dans quelle mesure le modèle peut être employé pour la prise de décision.
L’estimation directe du risque espéré par le risque empirique (estimation in-sample) serait excessivement optimiste pour des familles \(\mathcal{F}\) de capacité élevée (ou infinie) et doit donc être évitée. Lorsque la capacité de \(\mathcal{F}\) est faible et le nombre \(N\) de données d’apprentissage très élevé, des bornes de généralisation \(B(N,\mathcal{F})\) suffisamment réduites pour majorer de façon utile le risque espéré peuvent éventuellement être obtenues : \(R(f^*_{\mathcal{D}_N}) \leq R_{\mathcal{D}_N}(f^*_{\mathcal{D}_N}) + B(N,\mathcal{F})\). Cette situation est toutefois rare, même pour des données massives.
Une méthode générale pour estimer le risque espéré est celle de l’échantillon-test (estimation out-of-sample) :
L’ensemble de données disponibles \(\mathcal{D}_N\) est partitionné en deux ensembles mutuellement exclusifs par sélection aléatoire, les données d’apprentissage \(\mathcal{A}\) et les données de validation \(\mathcal{V}\).
L’apprentissage du modèle est réalisé sur les données de \(\mathcal{A}\) en utilisant une des approches mentionnées la MRE ou la MRER.
Le risque espéré du modèle résultant est estimé sur les données de \(\mathcal{V}\) (estimation out-of-sample).
Apprendre sur une partie seulement des données disponibles (\(\mathcal{A}\)) peut être un inconvénient sérieux lorsque \(N\) est faible. Même pour les données massives, certaines classes (ou certains comportements intéressants) peuvent être relativement rares dans les données, diminuer encore le nombre d’observations appartenant à ces classes (ou présentant ces comportements) a un impact négatif sur la qualité du modèle résultant.
Aussi, l’estimateur ainsi obtenu pour le risque espéré a une variance élevée (un autre découpage de \(\mathcal{D}_N\) en \(\mathcal{A}'\) et \(\mathcal{V}'\) peut produire un résultat assez différent). Pour réduire la variance de l’estimateur il faudrait moyenner les résultats issus de plusieurs découpages différents de \(\mathcal{D}_N\), c’est ce que proposent les méthodes de validation croisée.
Avant d’examiner de plus près ces méthodes, il est important de noter qu’un problème de validité important rencontré dans la pratique est la non stationnarité : les données d’apprentissage deviennent de moins en moins représentatives au fil du temps car la loi conjointe inconnue \(P\) évolue. Il est donc important d’estimer régulièrement l’erreur sur des données récentes afin de mettre en évidence une éventuelle divergence du modèle et donc la nécessité de le mettre à jour.
Afin de réduire la variance de l’estimation du risque espéré obtenue sur un échantillon-test, plusieurs partitionnements différents de \(\mathcal{D}_N\) en \(\mathcal{A}_i\) et \(\mathcal{V}_i\) sont réalisés, avec \(i \in {1,\ldots,k}\), \(\mathcal{D}_N = \mathcal{A}_i \cup \mathcal{V}_i\) et \(\mathcal{A}_i \cap \mathcal{V}_i = \varnothing\), à chaque fois un modèle \(f_i\) est appris sur \(\mathcal{A}_i\), son erreur \(L(\mathcal{V}_i,f_i)\) est calculée sur \(\mathcal{V}_i\), et enfin le risque espéré est estimé par la moyenne \(\frac{1}{k} \sum_{i=1}^k L(\mathcal{V}_i,f_i)\).
Suivant les partitionnements réalisés, les méthodes peuvent être
Exhaustives, lorsque tous les partitionnements possibles (respectant certains effectifs) sont utilisés :
Leave\(p\)out : \(N-p\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et \(p\) pour la validation (\(\mathcal{V}_i\)), tous les \(C_N^p\) partitionnements possibles avec ces effectifs sont utilisés. Il est donc nécessaire d’apprendre \(C_N^p\) modèles différents, ce qui implique en général un coût excessif.
Leave one out : \(N-1\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et une seule pour la validation (\(\|\mathcal{V}_i\| = 1\)), il y a donc \(C_N^1 = N\) partitionnements possibles et il faut apprendre \(N\) modèles différents. Le coût reste excessif pour des données massives (\(N\) très élevé).
Non exhaustives :
k-fold : l’ensemble \(\mathcal{D}_N\) est partitionné en \(k\) parties, chaque modèle est évalué sur une des partitions et les \(k-1\) autres sont employées pour son apprentissage. Sont appris \(k\) modèles, chacun évalué sur une partition différente parmi les \(k\). Souvent \(k = 10\), mais le choix de \(k\) dépend des ressources disponibles car le coût augmente linéairement avec \(k\). La méthode leave one out peut être vue aussi comme un cas particulier de k-fold, pour \(k = N\).
Échantillonnage répété : un échantillon aléatoire de \(n\) données est utilisé pour la validation, les autres \(N-n\) données étant employées pour l’apprentissage du modèle, on répète cela \(k\) fois pour obtenir \(k\) modèles. Cette méthode permet d’éviter la contrainte \(k \cdot n = N\), mais présente le désavantage de trouver certaines données dans plusieurs ensembles de validation différents alors que d’autres ne seront présentes dans aucun échantillon.
Même les méthodes non exhaustives sont coûteuses, car il est nécessaire d’apprendre (et ensuite évaluer) \(k\) modèles plutôt qu’un seul. Il faut toutefois noter que ces \(k\) apprentissages (et les \(k\) évaluations correspondantes) peuvent être réalisées en parallèle.
L’estimateur ainsi obtenu pour le risque espéré est asymptotiquement (lorsque \(k = N\) tend vers l’infini) sans biais. Pour \(k = N\) fini, cet estimateur présente néanmoins un léger biais, il surestime le risque espéré car l’apprentissage se fait à chaque fois sur moins de \(N\) données. Le biais sera ainsi plus faible pour leave one out que pour k-fold. La variance de l’estimateur reste relativement élevée, en partie à cause de l’utilisation de relativement peu de données pour évaluer chaque modèle. On considère que la validation croisée présente un bon compromis entre le biais et la variance de l’estimateur.
Une lecture attentive aura permis de remarquer que, pour répondre à l’objectif d’estimation du risque espéré pour un modèle, nous avons choisi d’en développer (et évaluer) \(k\). Pour lequel de ces modèles est valide cette estimation du risque espéré ? En général, pour traiter un problème de modélisation, avec un même ensemble \(\mathcal{D}_N\) sont employées plusieurs « procédures » de modélisation, chacune définie par une famille \(\mathcal{F}\), un critère de régularisation \(\textrm{Reg}(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc. La validation croisée sert ensuite à choisir, entre ces « procédures » de modélisation, celle pour laquelle le risque espéré estimé a la valeur la plus faible. La variance de l’estimateur étant élevée, ce choix n’est pas nécessairement optimal. La « procédure » choisie est ensuite employée pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). C’est à ce modèle que s’applique l’estimation du risque espéré obtenue pour la « procédure » dont le modèle est le résultat. Nous reviendrons plus loin sur les questions de sélection de modèle (ou de « procédure » de modélisation).
Problématique de l’échelle en apprentissage supervisé¶
La complexité du processus de construction de modèle par apprentissage supervisé et la complexité de la prise de décision avec un tel modèle peuvent varier suivant la famille \(\mathcal{F}\) et la méthode de modélisation considérée. La complexité fait partie des critères de choix, surtout dans le cas des données massives.
Considérons que la modélisation doit se faire à partir des \(N\) données de l’ensemble \(\mathcal{D}_N\) où chaque observation est décrite par \(p\) variables explicatives. Une fois fixée la famille paramétrique \(\mathcal{F}(\mathbf{w})\) de modèles, la complexité de l’algorithme d’optimisation permettant de déterminer le (vecteur de) paramètre(s) optimal \(\mathbf{w}^*\) peut être :
Pour les méthodes linéaires (régression linéaire, régression logistique, SVM linéaires), en général \(O(N \times p)\) car le nombre de paramètres est de \(O(p)\).
Pour les réseaux de neurones comme les perceptrons multicouches (non linéaires), \(O(N \times p^2)\) car le nombre de paramètres est de \(O(p^2)\) (avec \(p\) ou \(O(p)\) entrées et \(O(p)\) neurones dans une couche cachée, il y a \(O(p^2)\) connexions).
Pour les machines à vecteurs de support à noyaux non linéaires en général, la résolution directe du problème d’optimisation quadratique sous contraintes d’inégalité a une complexité de \(O(N^3 \times p)\), mais des méthodes de complexité \(O(N^2 \times p)\) sont couramment employées. Si l’espace d’arrivée est de dimension finie \(d\) et la fonction de transposition (feature map) de l’espace de départ vers l’espace d’arrivée, associée au noyau choisi, peut être explicitée, il peut être préférable de faire directement la modélisation linéaire dans l’espace d’arrivée, la complexité résultante sera \(O(N \times d)\).
Il faut également noter ici que certaines méthodes d’optimisation sont plus efficacement parallélisables que d’autres.
Avec le modèle \(f(\mathbf{w}^*)\) obtenu, la complexité de chaque prise de décision pour une nouvelle donnée \(\mathbf{x}\) peut être :
Pour les méthodes linéaires \(O(p)\), le nombre de paramètres étant de \(O(p)\).
Pour les perceptrons multicouches \(O(p^2)\), le nombre de paramètres étant de \(O(p^2)\).
Pour les machines à vecteurs de support à noyaux non linéaires, \(O(v \times p)\), où \(v\) est le nombre de vecteurs de support (on peut avoir \(v \propto N\)).
Nous examinons dans la suite les machines à vecteurs de support (SVM) et les solutions adoptées pour leur apprentissage lorsque \(N\) est élevé.
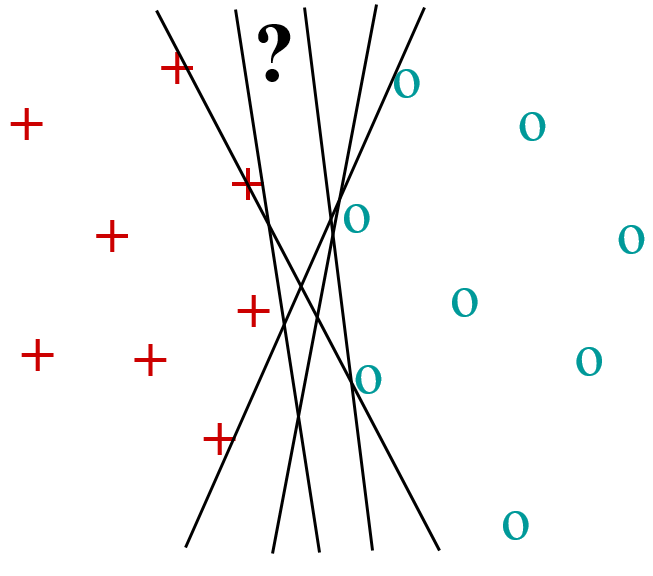
Considérons d’abord un problème de discrimination entre deux classes linéairement séparables, c’est à dire séparables parfaitement par une frontière linéaire. La figure suivante présente un exemple avec des données de \(\mathbb{R}^2\). Les données de \(\mathcal{D}_N\) sont les points rouges (classe 1) et bleus (classe 2). Dans cet exemple, une infinité de droites différentes séparent parfaitement les données de \(\mathcal{D}_N\) appartenant aux deux classes. Parmi ces droites, laquelle préférer comme modèle et pourquoi ?
Fig. 69 Quelle frontière pour séparer les classes ?¶
L’analyse discriminante linéaire propose une solution à ce problème, sur la base de la règle de décision bayésienne et le respect d’hypothèses de normalité et de homoscédasticité des classes (chaque classe est issue d’une loi normale multidimensionnelle et les matrices de variances-covariances sont identiques pour toutes les classes). Ces hypothèses étant rarement satisfaites, il est utile de chercher d’autres approches.
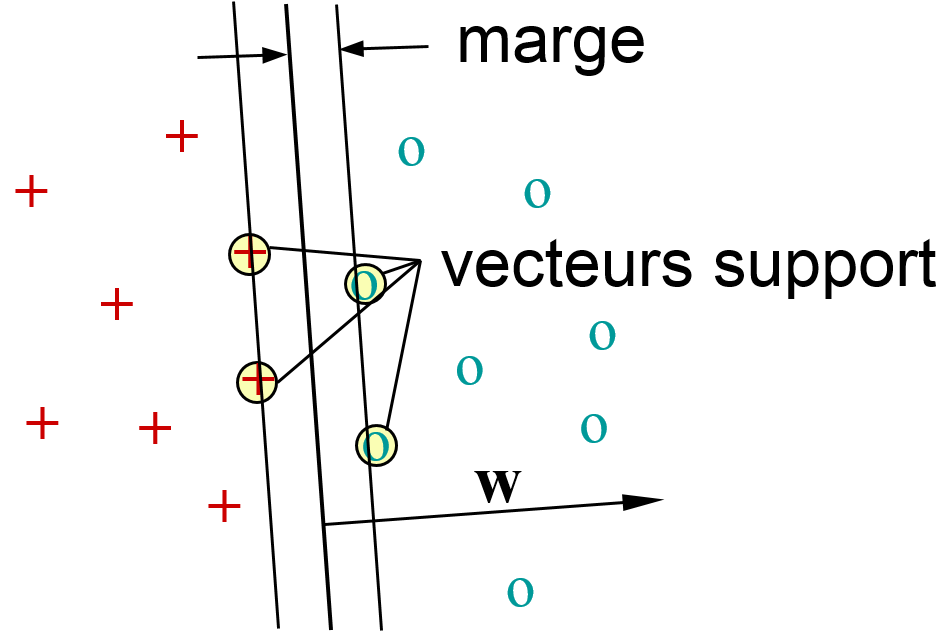
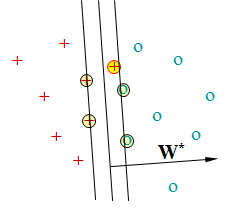
Les machines à vecteurs de support (Support Vector Machines, SVM, introduites par Vapnik, voir par ex. [ScS02]) proposent plutôt de préférer la frontière (le modèle) qui maximise la marge. Par marge on entend la distance entre la frontière et les données d’apprentissage les plus proches de chaque classe. Dans l’illustration suivante, la frontière qui maximise la marge est la droite située entre deux autres droites parallèles qui marquent le « bord » de chaque classe. Il est très important de noter que la position de cette frontière dépend exclusivement (pour des classes linéairement séparables) des données de \(\mathcal{D}_N\) situées sur ces « bords » de classes. Ces données sont appelées « vecteurs (de) support ». Dans l’illustration, ce sont les quatre points entourés.
En français on emploie fréquemment la traduction « séparateurs à vastes marges », qui a l’avantage de présenter les mêmes initiales SVM. Nous avons préféré ici la traduction « machines à vecteurs (de) support » car les SVM ne sont pas les seuls séparateurs à vastes marges et d’ailleurs les SVM ne sont pas uniquement des « séparateurs », vu qu’il est possible de s’en servir aussi pour la régression.
Pourquoi chercher à maximiser la marge ? Deux justifications sont en général mises en avant :
Les SVM sont issus des travaux théoriques de Vapnik et Chervonenkis sur une mesure de « capacité » de \(\mathcal{F}\) qui porte le nom de dimension de Vapnik-Chervonenkis (VC-dimension). On peut montrer que, pour des données vectorielles à l’intérieur d’une hypersphère de rayon \(r\), la VC-dimension \(h\) de la famille \(\mathcal{F}_m\) de hyperplans de marge \(\frac{1}{\|\mathbf{w}\|} \geq m\) est bornée, \(h \leq \left(\frac{r}{m}\right)^2 + 1\) ; noter que cette borne ne dépend pas de la dimension des données vectorielles considérées ! Or, l’écart entre le risque empirique et le risque espéré d’un modèle \(f \in \mathcal{F}_m\) est borné par \(B(N,h)\) qui diminue quand \(h\) diminue. Augmenter la marge permet donc de réduire \(h\) et donc de diminuer la borne \(B(N,h)\), donc d’atteindre de meilleures performances de généralisation.
Le risque espéré peut être estimé en utilisant la validation croisée leave one out. Or, nous avons remarqué que pour une SVM la frontière de discrimination dépendait exclusivement des vecteurs support. Lorsque la validation croisée leave one out est appliquée, un modèle est appris sur toutes les données sauf une et est ensuite évalué sur la donnée mise de côté. Si cette donnée n’est pas un vecteur support de l’apprentissage avec toutes les données, le modèle résultant reste le même et classe bien la donnée mise de côté (erreur nulle). En revanche, si cette donnée est un vecteur support de l’apprentissage avec toutes les données, le modèle résultant est probablement différent et peut classer mal (ou non) la donnée mise de côté. Si \(v\) est le nombre de vecteurs support, sur les \(N\) modèles différents obtenus par validation croisée leave one out, au maximum \(v\) font une erreur lors de leur évaluation (sur la donnée exclue de leurs données d’apprentissage). On peut ainsi montrer que le risque espéré est borné par le rapport entre le nombre de vecteurs support et le nombre total de données d’apprentissage, \(R(f) \leq \frac{E[v(N)]}{N}\), où \(E[v(N)]\) est l’espérance du nombre de vecteurs support avec \(N\) données d’apprentissage. Cette borne est surtout intéressante si \(v \ll N\). Un tel résultat est rendu possible par le choix de maximiser la marge. Cet aspect est approfondi dans [Joa99], où sont proposés d’autres estimateurs de l’erreur de généralisation pour les SVM.
SVM : problème d’optimisation pour une séparation linéaire¶
Une frontière de séparation linéaire est, dans un cas général, un hyperplan défini de façon unique par un vecteur normal \(\mathbf{w}\) (pour son « orientation ») et une constante \(b\) (pour sa position exacte) : un vecteur \(\mathbf{x}\) est situé sur l’hyperplan si et seulement si \(\mathbf{w}^T \mathbf{x} + b = 0\).
Si les données de \(\mathcal{D}_N\) sont linéairement séparables, il est possible de trouver des hyperplans tels que les données \((\mathbf{x}_i,y_i) \in \mathcal{D}_N\) appartenant à une des classes (par ex. \(y_i = 1\)) soient d’un côté de l’hyperplan (\(\mathbf{w}^T \mathbf{x}_i + b > 0\)) et les données appartenant à l’autre classe ((par ex. \(y_i = -1\)) de l’autre côté de l’hyperplan (\(\mathbf{w}^T \mathbf{x}_i + b < 0\)). Parmi les hyperplans qui satisfont cette condition, il faut identifier celui qui maximise la marge, caractérisé par \(\mathbf{w}^*\) et \(b^*\).
On observe que l’équation d’un hyperplan, \(\mathbf{w}^T \mathbf{x}_i + b = 0\), reste satisfaite pour \(\mathbf{w} \rightarrow k \mathbf{w}\), \(b \rightarrow k b\), avec \(k \in \mathbb{R}\). Pour que le problème de détermination de \(\mathbf{w}^*\), \(b^*\) soit bien posé, il faut donc ajouter une condition de normalisation : pour tout vecteur de support \(\mathbf{x}_{SV}\), on impose \(\left|\mathbf{w}^T \mathbf{x}_{SV} + b\right| = 1\). Avec cette condition, on constate que la marge est \(\frac{1}{\|\mathbf{w}\|}\).
La recherche de l’hyperplan qui maximise la marge revient alors à un problème d’optimisation sous contraintes d’inégalité :
(12)¶\[\begin{split}\Bigg\{
\begin{array}{l}
\min_{\mathbf{w},b} \frac{1}{2}\|\mathbf{w}\|^2\\
y_i \left(\mathbf{w}^T \mathbf{x}_i + b\right) \geq 1, \, 1 \leq i \leq N
\end{array}\end{split}\]
La condition d’optimisation correspond à la maximisation de la marge (\(= \frac{1}{\|\mathbf{w}\|}\)), les inégalités indiquent que seuls les vecteurs de support (non connus au départ, identifiés par le processus d’optimisation) se situent sur les « bords » des classes, \(y_i \left(\mathbf{w}^T \mathbf{x}_{SV} + b\right) = 1\), les autres données de \(\mathcal{D}_N\) doivent être au-delà (plus loin de l’hyperplan recherché), \(y_i \left(\mathbf{w}^T \mathbf{x}_i + b\right) > 1\).
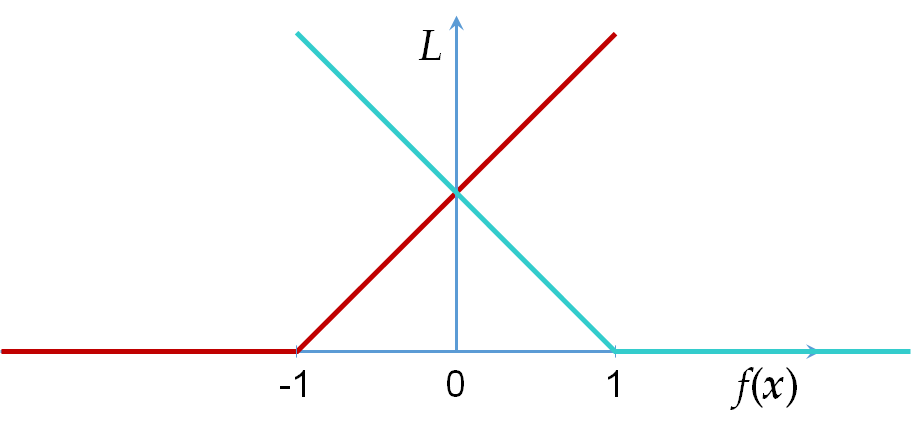
Les résultats obtenus avec une séparation linéaire peuvent être suffisamment bons même lorsque les classes ne sont pas tout à fait linéairement séparables. Pour trouver le meilleur modèle linéaire avec des données de \(\mathcal{D}_N\) qui ne sont pas linéairement séparables, il est nécessaire d’utiliser une fonction d’erreur (de perte) adaptée. La fonction hinge loss, \(L_H(\mathbf{x},y,f) = \max\{0, 1 - y f(\mathbf{x})\}\), \(y \in \{-1;1\}\), illustrée dans la figure suivante, est bien adaptée.
L’application de cette fonction pour les données des deux classes est représentée dans la figure suivante. Tant qu’une donnée reste du bon côté du « bord » de sa classe, l’erreur est nulle ; lorsqu’une donnée est du mauvais côté du « bord » de sa classe, l’erreur augmente linéairement avec l’éloignement de ce « bord » :
Fig. 72 Hinge loss appliquée aux données de chaque classe¶
Le modèle recherché correspondra à un compromis entre la maximisation de la marge et la minimisation du nombre de données de \(\mathcal{D}_N\) se trouvant du mauvais côté des « bords » des classes (et leur éloignement du bord correspondant). Il est nécessaire d’introduire un paramètre pour pondérer un de ces deux critères par rapport à l’autre. Le critère à optimiser employé sera :
(13)¶\[\begin{split}\Bigg\{
\begin{array}{l}
\min_{\mathbf{w},b} \left(\frac{1}{2}\|\mathbf{w}\|^2 + C \sum_{i=1}^N \xi_i \right)\\
y_i \left(\mathbf{w}^T \mathbf{x}_i + b\right) \geq 1 - \xi_i, \,\, \xi_i \geq 0, \,\, 1 \leq i \leq N
\end{array}\end{split}\]
où \(\xi_i\) correspond à l’erreur faite pour la donnée \(i\) de \(\mathcal{D}_N\) et \(C\) est la pondération de l’erreur totale dans la fonction à minimiser. \(C\) est considéré comme un paramètre de régularisation, car il permet de contrôler l’importance donnée à la maximisation de la marge (qui vise à reduire la capacité de la famille \(\mathcal{F}\) de modèles). Il faut toutefois noter que \(C\) pondère l’erreur et non le terme de régularisation : plus la valeur de \(C\) est élevée, plus le poids de la minimisation de l’erreur d’apprentissage est élevé par rapport à celui de la maximisation de la marge (et donc plus faible est la régularisation).
Bien entendu, ce critère combiné peut être utilisé même lorsque les données de \(\mathcal{D}_N\)sont linéairement séparables car cela permet d’élargir la marge, quitte à ce que certaines des données de \(\mathcal{D}_N\) se trouvent du mauvais côté des « bords » des classes ou même de la frontière de séparation, comme dans la figure suivante :
La résolution du problème d’optimisation permet de déterminer le modèle, caractérisé par \(\mathbf{w}^*\) et \(b^*\). La fonction de décision correspondante, appliquée à une donnée (observation) \(\mathbf{x}\), est :
Cette fonction de décision s’annule sur l’hyperplan qui est la frontière de discrimination. L’observation \(\mathbf{x}\) est affectée à une des classes suivant le signe de la fonction de décision.
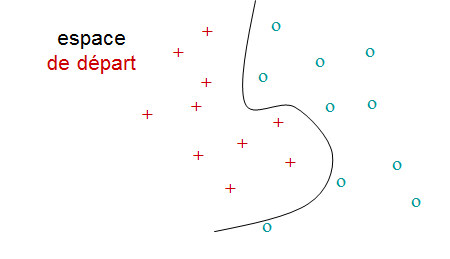
Comment procéder si une séparation linéaire des classes est vraiment inadaptée (l’erreur obtenue est trop élevée), comme dans l’exemple de la figure suivante ?
La solution consiste à transposer les données dans un autre espace, de dimension supérieure (éventuellement infinie), dans lequel une séparation linéaire donne de bons résultats, comme dans la figure suivant :
A chaque donnée \(\mathbf{x}\) de l’espace de départ, par ex. \(\mathbb{R}^p\), la fonction de transposition (feature map) \(\phi : \mathbb{R}^p \rightarrow \mathcal{H}\) associe une image \(\phi(\mathbf{x})\) dans l’espace « d’arrivée » \(\mathcal{H}\).
Définir et résoudre le problème d’optimisation (13), appelé problème primal, dans l’espace d’arrivée \(\mathcal{H}\) peut être impossible (par ex. si la fonction \(\phi\) ne peut pas être exprimée sous une forme analytique) ou peu pratique (par ex. si la dimension de l’espace d’arrivée n’est pas finie). Heureusement, si la fonction de transposition \(\phi\) peut être associée à une fonction noyau \(K\)valide (nous expliciterons cette notion dans la section suivante) par la relation \(\left\langle \phi(\mathbf{x}_i),\phi(\mathbf{x}_j) \right\rangle = K(\mathbf{x}_i,\mathbf{x}_j)\), \(\forall \mathbf{x}_i,\mathbf{x}_j\), où \(\left\langle \cdot,\cdot \right\rangle\) est un produit scalaire dans \(\mathcal{H}\), alors tous les calculs impliquant des produits scalaires dans l’espace d’arrivée \(\mathcal{H}\) peuvent être exprimés par des calculs de noyau \(K\) dans l’espace de départ. Or, le problème d’optimisation (13) fait appel uniquement à de tels produits scalaires. On appelle cela l”« astuce du noyau » (kernel trick).
Le problème d’optimisation primal dans l’espace d’arrivée est ainsi traduit, par l’emploi de la fonction noyau, en un problème d’optimisation quadratique dual dans l’espace de départ :
Le modèle résultant de l’algorithme d’optimisation est défini par les coefficients \(\alpha_i^*, 1 \leq i \leq N\) et \(b^*\). Seuls les vecteurs de support ont des coefficients \(\alpha_i^*\) non nuls.
La fonction de décision correspondante appliquée à une donnée \(\mathbf{x}\) est, lorsqu’on l’écrit en utilisant le noyau :
Par noyau « valide » on entend un noyau qui garantit l’existence d’un espace d’arrivée \(\mathcal{H}\) et de la fonction de transposition \(\phi\) tels que \(K(\mathbf{x}_i,\mathbf{x}_j) = \left\langle \phi(\mathbf{x}_i),\phi(\mathbf{x}_j) \right\rangle\), \(\left\langle \cdot,\cdot \right\rangle\) étant le produit scalaire de \(\mathcal{H}\). Pour des données vectorielles, le résultat suivant permet de définir une condition suffisante.
Soit \(\mathcal{X}\) compact dans \(\mathbb{R}^p\) et \(K : \mathcal{X} \times \mathcal{X} \rightarrow \mathbb{R}\), symétrique, tel que \(\forall f \in L_2(\mathcal{X})\), \(\int_{\mathcal{X}^2} K(\mathbf{x},\mathbf{y}) f(\mathbf{x}) f(\mathbf{y}) d\mathbf{x} d\mathbf{y} \geq 0\) (noyau défini positif[1], ou condition de Mercer). Alors il existe un espace de Hilbert \(\mathcal{H}\) et \(\phi : \mathcal{X} \rightarrow \mathcal{H}\) tels que \(K(\mathbf{x}_i,\mathbf{x}_j) = \left\langle \phi(\mathbf{x}_i),\phi(\mathbf{x}_j) \right\rangle\), \(\forall \mathbf{x}_i,\mathbf{x}_j\), où \(\left\langle \cdot,\cdot \right\rangle\) est le produit scalaire de \(\mathcal{H}\). L’existence de \(\mathcal{H}\) et de \(\phi\) étant ainsi garanties, il n’est pas nécessaire de connaître \(\mathcal{H}\) ou d’exprimer \(\phi\) pour pouvoir faire les calculs de (15) et (16) en employant \(K\).
Différents noyaux valides sont couramment employés pour des données vectorielles : le noyau linéaire, le noyau gaussien (ou RBF), noyaux polynomiaux, le noyau angulaire (conditionnellement défini positif), etc. Pour certains noyaux (polynomiaux, par ex.), la fonction \(\phi\) peut être explicitée et les calculs facilement réalisés dans l’espace d’arrivée, il peut donc être préférable de résoudre directement le problème d’optimisation primal dans l’espace d’arrivée.
Un intérêt majeur des SVM est la possibilité de travailler directement sur des données non vectorielles (par ex. séquences de longueur variable, arbres, graphes) en choisissant un noyau adapté au type de données. L’espace de Hilbert \(\mathcal{H}\), la fonction de transposition \(\phi\) et le noyau \(K(\cdot,\cdot) = \left\langle \phi(\cdot,\cdot) \right\rangle\) sont souvent construits explicitement. Voir par exemple [Gär03] pour un passage en revue de noyaux pour données structurées.
Enfin, il est possible de construire des noyaux valides en combinant d’autres noyaux valides, par exemple pour obtenir des noyaux pour données hybrides (décrites par plusieurs variables de nature différente) ; on parle de l”« ingénierie des noyaux ». Quelques détails supplémentaires sur les SVM pour la discrimination, pour la régression, pour l’estimation du support d’une distribution, ainsi que sur l’analyse factorielle à noyaux et sur l’ingénierie des noyaux peuvent être trouvés par ex. ici.
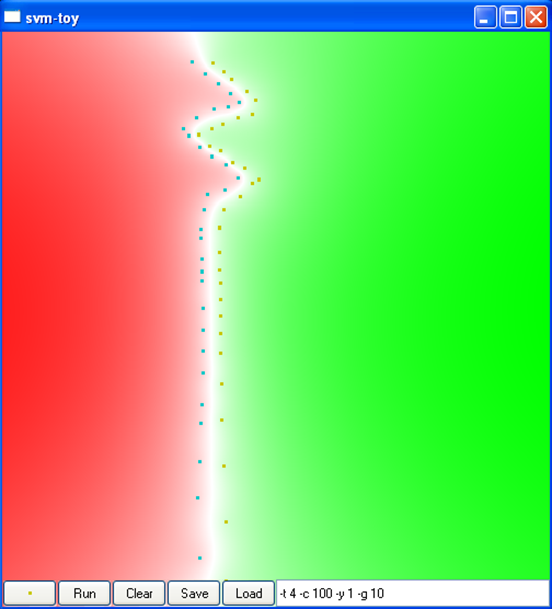
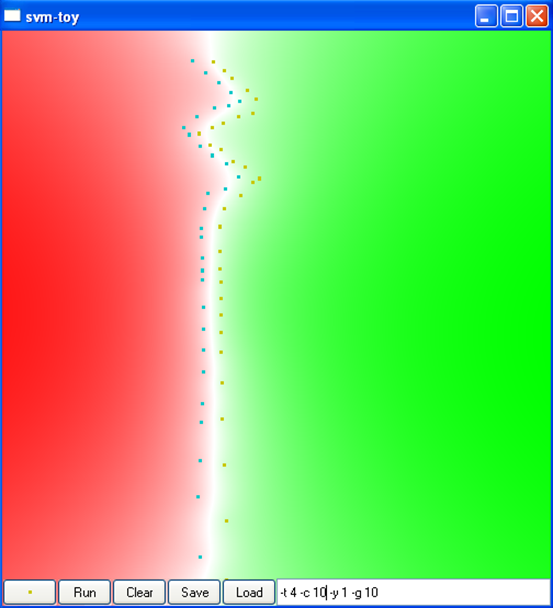
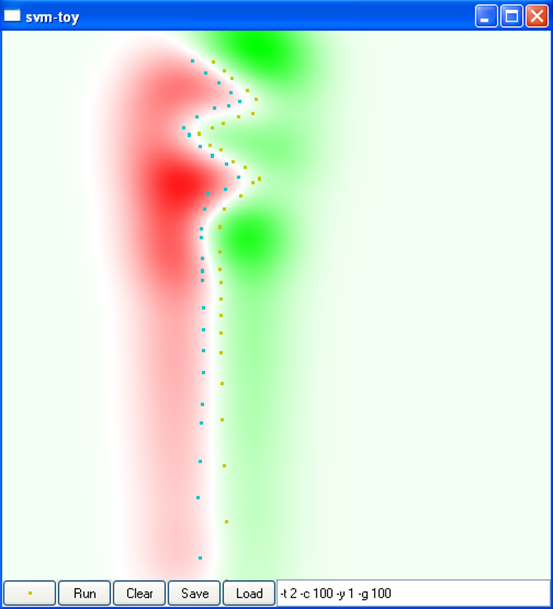
La figure suivante montre l’impact de la variance du noyau RBF sur la discrimination obtenue dans un problème à deux classes dans \(\mathbb{R}^2\), ainsi que sur le nombre de vecteurs de support. L’intérieur de claque classe est représenté en rouge et respectivement en vert, la frontière de séparation est représentée en blanc (cliquez sur une image pour la voir plus grande) :
Par « sur-apprentissage » on entend un cas où le risque empirique (l’erreur d’apprentissage) est faible mais le risque espéré (l’erreur de généralisation) élevé. Par « sous-apprentissage » on entend un cas où le risque empirique reste élevé. On peut constater que la variance du noyau RBF a un impact très significatif sur les résultats.
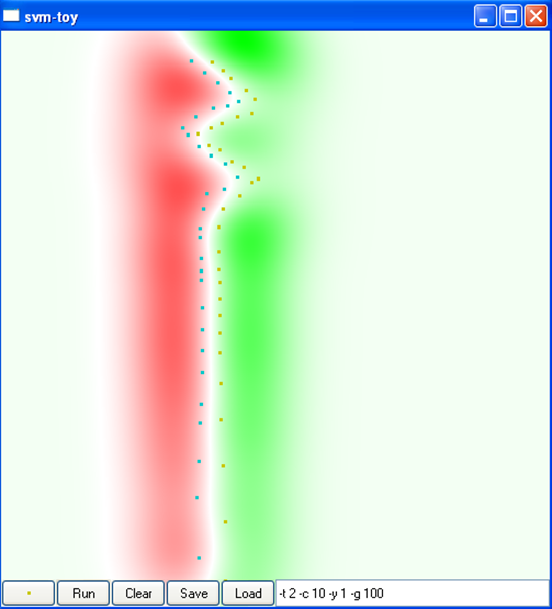
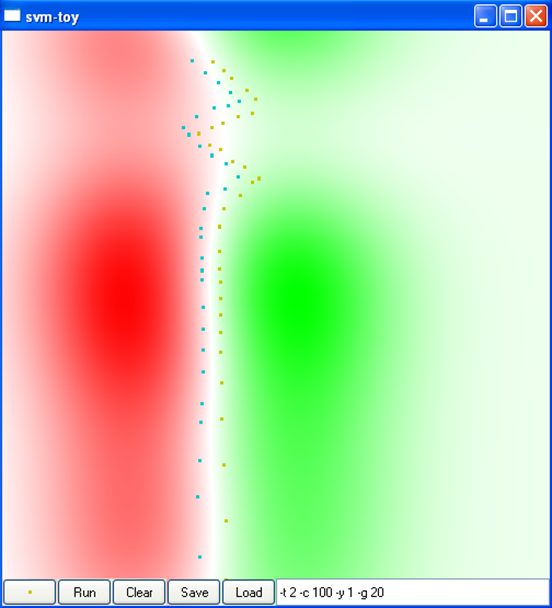
La figure suivante montre l’impact du type de noyau, de la variance du noyau (pour le noyau RBF, car le noyau angulaire n’a pas de paramètres) et de la constante \(C\) sur la discrimination obtenue dans un problème à deux classes dans \(\mathbb{R}^2\) (cliquez sur une image pour la voir plus grande) :
Noyau angulaire
Noyau RBF
C = 100
C = 10
Var. 0,01, C = 100
Var. 0,01, C = 10
Noyau RBF
Noyau RBF
Var. 0,05, C = 100
Var. 0,01, C = 100
Var. 0,001, C = 100
Var. 0,001, C = 0,7
Une valeur élevée de \(C\) accorde plus de poids à la minimisation du risque empirique et force ainsi le modèle résultant à suivre de plus près les « détails » de la séparation entre les classes sur les données d’apprentissage. On constate aussi qu’il est nécessaire de tenir compte de plusieurs « hyperparamètres » (type du noyau, variance du noyau, constante \(C\)) pour rechercher la meilleure solution. Nous y reviendrons dans la section Hyperparamètres et sélection de modèle plus loin.
L’apprentissage des SVM linéaires est réalisé par la résolution du problème d’optimisation (13) dans l’espace de départ avec par ex. la descente de (sous-)gradient stochastique, facilement parallélisable, comme nous le verrons plus loin.
Lorsque le problème exige des SVM (à noyaux) non linéaires, plusieurs possibilités existent :
Si le noyau permet de déterminer une représentation vectorielle explicite et de dimension finie dans l’espace d’arrivée (c’est le cas, par ex., pour les noyaux polynomiaux), il est possible d’exprimer et de résoudre le problème d’optimisation primal(13)dans l’espace d’arrivée. Les algorithmes de résolution des modèles linéaires peuvent alors être employés, avec leurs avantages de complexité linéaire en \(N\) et de parallélisation facile.
Pour les noyaux qui ne présentent pas ces caractéristiques (par ex. le noyau RBF), il faut résoudre le problème dual(15) qui exige des solveurs quadratiques dont les opérations ne sont pas parallélisables de façon efficace. Pour passer à l’échelle, différentes pistes sont explorées (voir également [BCDW07]) :
L’approximation du noyau original par un noyau permettant d’expliciter une représentation d’arrivée de dimension finie. Les résultats sont néanmoins différents de ceux qui seraient obtenus avec le noyau original.
L’emploi de méthodes de réduction de la complexité pour noyaux à décroissance rapide (par ex. RBF), comme indiqué dans le paragraphe sur l’apprentissage supervisé dans le cours sur la réduction de l’ordre de complexité. Cela est intéressant si la variance du noyau employé est faible par rapport à l’étendue du domaine couvert par les données de \(\mathcal{D}_N\).
Plus généralement, pour réduire le coût de l’apprentissage supervisé (par SVM ou autre méthode) il est envisageable d’effectuer cet apprentissage en plusieurs étapes successives et, lors de chaque étape, sur un volume de données comparativement faible. Cela est possible (entre autres) par :
Apprentissage incrémental : le principe est de réaliser un premier apprentissage sur un échantillon de \(\mathcal{D}_N\), ensuite un deuxième apprentissage sur la réunion entre un nouvel échantillon de \(\mathcal{D}_N\) et les vecteurs de support issus du premier apprentissage, et ainsi de suite jusqu’à épuisement de \(\mathcal{D}_N\) ou absence de changement dans l’ensemble de vecteurs de support. Ainsi, à chaque itération on apprend sur un faible volume de données, à condition que le nombre de vecteurs de support issus de l’itération précédente soit bien plus faible que la taille de l’échantillon.
Apprentissage actif : le principe est de faire un apprentissage initial sur un échantillon de \(\mathcal{D}_N\), ensuite d’ajouter progressivement, lors d’itérations successives, les données de \(\mathcal{D}_N\) qui peuvent améliorer le plus le modèle courant.
Pour une machine à vecteurs de support, la complexité de la prise de décision pour une nouvelle donnée \(\mathbf{x}\) est \(O(v \times p)\), \(v\) étant le nombre de vecteurs de support (qui peut être \(v \propto N\)). Pour réduire le coût de la prise de décision lorsque \(v\) est très élevé, on peut envisager :
L’approximation de la solution avec nettement moins de vecteurs support, voir par ex. [DT05].
L’application de méthodes de réduction de la complexité pour noyaux à décroissance rapide, comme indiqué dans le paragraphe sur la prise de décision dans le cours sur la réduction de l’ordre de complexité.
SVM linéaires et descente de gradient stochastique¶
Pour les SVM linéaires, dans le problème d’optimisation à résoudre, donné par (13), le terme à minimiser peut être écrit sous la forme suivante :
\[\min_{\mathbf{w},b} \left(\frac{1}{2}\|\mathbf{w}\|^2 + C \sum_{i=1}^N L_H(\mathbf{x}_i,y_i;\mathbf{w},b) \right)\]
où \(L_H(\mathbf{x}_i,y_i;\mathbf{w},b) = \max \{0, 1 - y_i \left(\mathbf{w}^T \mathbf{x}_i + b\right)\}\) est le hinge loss. Il est possible d’inclure le seuil \(b\) comme composante supplémentaire de \(\mathbf{w}\), en ajoutant une composante supplémentaire correspondante, toujours égale à \(1\), à l’entrée \(\mathbf{x}\). Le problème d’optimisation devient \(\min_{\mathbf{w}} g(\mathbf{w})\), où
\[g(\mathbf{w}) = \left(\frac{1}{2}\|\mathbf{w}\|^2 + C \sum_{i=1}^N L_H(\mathbf{x}_i,y_i;\mathbf{w}) \right)\]
avec \(L_H(\mathbf{x}_i,y_i;\mathbf{w}) = \max \{0, 1 - y_i (\mathbf{w}^T \mathbf{x}_i)\}\).
Pour minimiser une fonction différentiable \(l(\mathbf{w})\), une solution simple est la descente de gradient :
Notre fonction \(g\) n’est pas différentiable partout car le hinge loss n’est pas différentiable pour \(y_i (\mathbf{w}^T \mathbf{x}_i) = 1\). Il est en revanche possible d’employer pour \(g\) la descente de sous-gradient, suivant la même équation que (17) mais où le gradient est remplacé par le sous-gradient calculé suivant :
Minimisation de l’inverse de la marge (terme différentiable) :
De plus, la composante \(\sum_{i=1}^N L_H(\mathbf{x}_i,y_i;\mathbf{w})\) étant additive par rapport aux données \((\mathbf{x}_i,y_i)\), une version stochastique de la descente de sous-gradient peut être définie : à chaque itération \(t\), un \(i\) est choisi et \(\mathbf{w}_{t+1}\) est mis à jour sur la base de la seule contribution de \(\frac{\partial}{\partial \mathbf{w}} L_H(\mathbf{x}_i,y_i;\mathbf{w}_t)\). On préfère souvent une variante mini-batch, dans laquelle un échantillon de faible taille est choisi à chaque itération plutôt qu’une seule donnée.
L’implémentation MapReduce de la descente de (sous-)gradient stochastique mini-batch est facile :
Entrées : ensemble \(\mathcal{D}_N\) de \(N\) données \((\mathbf{x}_i,y_i) \in \mathbb{R}^p \times \{-1;1\}\), taux d’échantillonnage \(s\), pas initial \(\eta\) ;
Sorties : Vecteur \(\mathbf{w}\) et seuil \(b\) ;
\(\mathcal{D}_N\) est découpé en fragments (partitions) distribués aux nœuds de calcul ; un fragment doit tenir dans la mémoire d’un nœud ;
Une solution initiale est choisie pour \(\mathbf{w}_0\) et \(b_0\) ;
tantque (nombre maximal d’itérations non atteint) faire
La solution courante \(\mathbf{w}_t\) et \(b_t\) est transmise à tous les nœuds de calcul ;
Chaque tâche Map choisit un échantillon de données \((\mathbf{x}_i,y_i)\) de son fragment, en respectant le taux d’échantillonnage \(s\) ;
Chaque tâche Map calcule la contribution de cet échantillon à \(\mathbf{w}_{t+1}\) et \(b_{t+1}\) ;
Les résultats des tâches Map sont transmis aux tâches Reduce qui calculent \(\mathbf{w}_{t+1}\) et \(b_{t+1}\) en ajoutant également la contribution du terme de maximisation de la marge ;
fintantque
Dans l’étape 3.4, le partitionnement pour les tâches Reduce peut être fait, par exemple, par groupe de dimensions de \(\mathbb{R}^p\).
Évaluation pour problèmes avec une classe d’intérêt¶
L’évaluation de base d’un modèle discriminant sur un ensemble de données \(\mathcal{E}\) est réalisée à travers le taux de mauvais classement des données de l’ensemble de test, qui correspond à l’erreur moyenne (sur les données de test) obtenue en utilisant la perte 0-1. Une telle évaluation est toutefois réductrice car la perte 0-1 fait l’hypothèse d’un coût de mauvais classement symétrique : le coût est égal à 1 quelle que soit l’erreur d’affectation. Or, dans de nombreux cas les erreurs d’affectation ne sont pas équivalentes. Par exemple, ne pas détecter la maladie grave d’un patient à partir de mesures cliniques est dramatique, alors que signaler une maladie grave pour un patient qui n’en est pas atteint oblige simplement à effectuer un ou plusieurs examens complémentaires. Bien entendu, si cette asymétrie peut être quantifiée alors elle peut être prise en compte dès le départ, dans la construction du modèle décisionnel. Il est toutefois utile de pouvoir comparer plusieurs modèles discriminants sans fixer a priori un « degré » d’asymétrie.
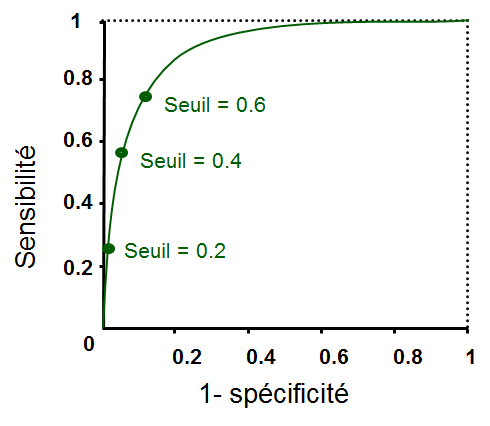
Les courbes ROC (initiales provenant de receiver operating characteristic, ou « caractéristique de fonctionnement du récepteur » en traduction littérale) répondent à cet objectif. On considère un problème de discrimination dans lequel une des classes est la « classe d’intérêt » et un modèle discriminant appris à partir des données est vu comme le « détecteur » de la classe d’intérêt.
Un modèle discriminant est en général décrit par un vecteur de paramètres (par ex. \(\mathbf{w}\), qui indique l’orientation de l’hyperplan de séparation pour une SVM linéaire) et un seuil de détection (\(b\), la position de l’hyperplan pour une SVM linéaire). Le principe d’une courbe ROC est de balayer la totalité du domaine utile de variation de ce seuil et d’examiner, pour chaque valeur du seuil, les résultats obtenus sur les données d’évaluation, représentés par la matrice de confusion :
Positifs (\(\in\) classe intérêt)
Négatifs (\(\notin\) classe intérêt)
Détectés positifs
Vrais Positifs
Faux Positifs
Détectés négatifs
Faux Négatifs
Vrais Négatifs
Chaque donnée de \(\mathcal{E}\) peut appartenir à la classe d’intérêt (Classe présente) ou à une autre classe (Classe [d’intérêt] absente) ; cette information est supposée connue pour toutes les données de \(\mathcal{E}\) (« vérité terrain »). Chaque donnée peut être affectée par le modèle à la classe d’intérêt (Classe détectée) ou à une autre classe (Classe non détectée). Si une donnée fait partie de la classe d’intérêt mais est affectée par le modèle à une autre classe, on parle d’un « faux négatif ». Si une donnée est affectée par le modèle à la classe d’intérêt alors qu’elle fait partie d’une autre classe, on parle d’un « faux positif ».
A partir du contenu d’une matrice de confusion, on définit les mesures suivantes :
La sensibilité, ou le taux de vrais positifs (true positive rate, TPR), mesure la capacité du modèle à détecter les vrais positifs. Si tous les vrais positifs sont détectés (s’il n’y a pas de faux négatifs, c’est à dire des positifs non détectés comme positifs), alors la sensibilité est égale à 1.
La 1 - spécificité, ou le taux de faux positifs (false positive rate, FPR), mesure la capacité du modèle à détecter seulement les vrais positifs. Si aucun négatif n’est détecté comme positif (s’il n’y a pas de faux positifs), alors la 1 - spécificité est égale à 0 (c’est à dire, la spécificité est égale à 1).
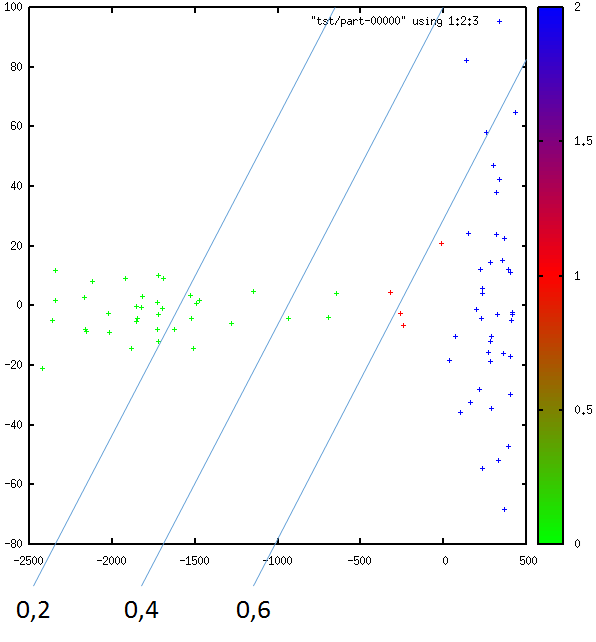
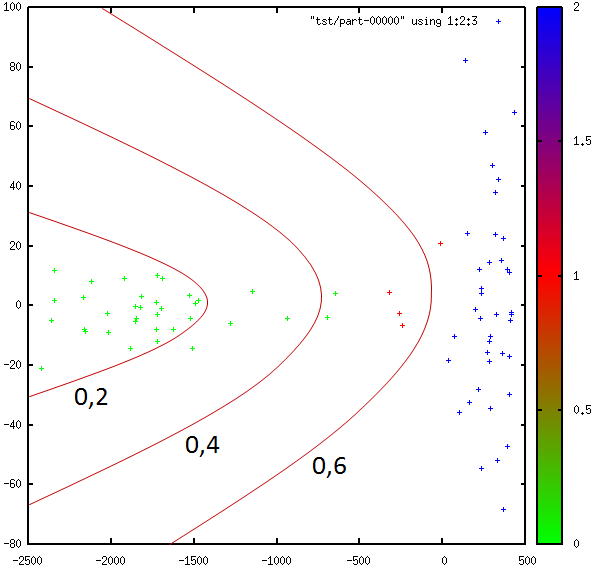
Dans la figure suivante sont représentées des données de \(\mathbb{R}^2\). Celles de la classe d’intérêt sont en vert et celles des autres classes en bleu. Considérons un modèle discriminant correspondant à une frontière de séparation verticale (le vecteur \(\mathbf{w}\), orthogonal à cette frontière, est horizontal). Les points rouges correspondent en partie à des données de la classe d’intérêt situées, par rapport à une frontière verticale, du côté des données des autres classes, et en partie à des données des autres classes situées du côté de la classe d’intérêt. Dans cet exemple, la classe d’intérêt n’est jamais parfaitement séparée des autres classes, quelle que soit la position de la frontière verticale.
Fig. 78 Positions de la frontière verticale de discrimination entre deux classes de \(\mathbb{R}^2\)¶
En explorant la totalité du domaine de variation utile du seuil (\(b\), pour une SVM linéaire) et en enregistrant à chaque fois la sensibilité et 1 - spécificité, on obtient le graphique suivant :
Lorsque la frontière est très à gauche, toutes le données sont du côté « négatif ». Aucun vrai positif n’est détecté et aucun négatif n’est détecté comme positif. La sensibilité est donc nulle et la 1 - spécificité est nulle également.
Au fur et à mesure que la frontière se déplace vers la droite, de plus en plus de positifs sont détectés mais aucun négatif n’est détecté comme positif. La sensibilité augmente et la 1 - spécificité reste nulle. A partir d’une valeur du seuil, des négatifs commencent à être détectés comme positifs. La sensibilité commence donc à augmenter. Si la frontière continue son déplacement vers la droite, à partir d’une autre valeur du seuil il n’y a plus aucun positif du mauvais côté de la frontière, tous sont détectés comme des positifs. La sensibilité atteint donc 1. En revanche, comme de plus en plus de négatifs sont détectés comme positifs, la 1 - spécificité augmente.
Lorsque la frontière est suffisamment à droite, toutes le données sont du côté « positif ». Tous les positifs sont détectés et tous les négatifs sont détectés comme positifs. La sensibilité est donc égale à 1 et la 1 - spécificité égale à 1 aussi.
Il est utile de considérer deux autres modèles discriminants et d’examiner leurs courbes ROC. Un premier modèle est linéaire aussi mais correspond à des frontières inclinées de \(30\,^{\circ}\)), comme dans la figure suivante :
Fig. 80 Positions de frontières obliques de discrimination entre deux classes de \(\mathbb{R}^2\)¶
Un second modèle est non linéaire, sur la région considérée de \(\mathbb{R}^2\) les frontières correspondantes ont la forme de segment de paraboles, comme dans la figure suivante :
Fig. 81 Positions de frontières non linéaires de discrimination entre deux classes de \(\mathbb{R}^2\)¶
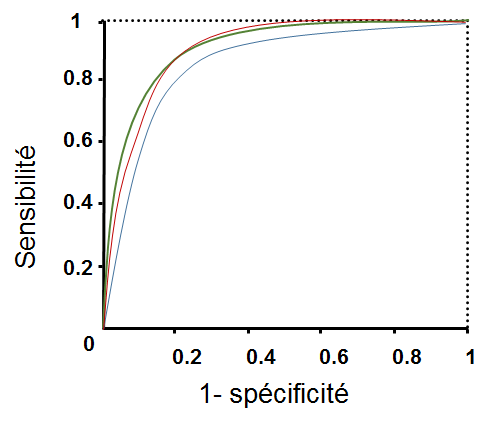
Peut-on comparer ces modèles à partir de leurs courbes ROC, pour constater éventuellement qu’un est plus intéressant (ou moins intéressant) que les autres sur la totalité du domaine de variation du seuil ?
La figure suivante montre que la courbe ROC du modèle oblique se situe systématiquement en-dessous des deux autres, en revanche les courbes ROC du modèle vertical et du modèle non linéaire se croisent. Une mesure couramment employée pour comparer deux modèles sur la totalité du domaine de variation utile du seuil est l’aire sous la courbe ROC, notée en général AUC (area under curve). Plus sa valeur est élevée, plus le modèle est intéressant (performant). Si deux modèles ont des valeurs AUC très proches, le choix doit être fait à partir d’autres critères, par exemple en comparant les sensibilités à spécificité fixée.
Fig. 82 Comparaison de modèles utilisant les courbes ROC¶
Le traitement d’un problème de modélisation décisionnelle à partir d’un ensemble de données \(\mathcal{D}_N\) passe en général par l’évaluation de plusieurs « procédures » de modélisation différentes, chacune définie par une famille de modèles \(\mathcal{F}\), un critère de régularisation \(\textrm{Reg}(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc. Ces « procédures » de modélisation sont ensuite comparées en utilisant la validation croisée afin de retenir celle pour laquelle le risque espéré (l’erreur de généralisation) estimé a la valeur la plus faible.
Les paramètres numériques dont dépend une procédure de modélisation sont en général appelés « hyperparamètres » (terme employé à l’origine pour les paramètres des distributions a priori dans les statistiques bayesiennes). Par exemple, sont considérés hyperparamètres :
Pour les SVM linéaires, la valeur de \(C\) dans (13) (appelé aussi paramètre de régularisation).
Pour les SVM à noyau RBF, la valeur de \(C\) dans (13), mais aussi la variance du noyau RBF employé. Nous avons vu plus haut, pour des exemples simples, l’impact de la valeur de \(C\) et de la variance du noyau sur les résultats obtenus.
Parfois, le terme « hyperparamètres » est étendu aux variables nominales définissant le famille de modèles \(\mathcal{F}\), comme par exemple le type de noyau employé pour des SVM.
Pour trouver le meilleur modèle décisionnel il est nécessaire de comparer plusieurs « procédures » de modélisation différentes. Chaque « procédure » de modélisation est définie par une valeur spécifique pour chaque hyperparamètre. Une fois fixées les valeurs des hyperparamètres (par ex. de \(C\) pour les SVM linéaires), l’algorithme d’optimisation (par ex. la descente de sous-gradient stochastique) opère pour déterminer le modèle (par ex. \(\mathbf{w}^*\) et \(b^*\) pour une SVM linéaire).
Les meilleures valeurs des hyperparamètres sont identifiées de la manière suivante :
Définition d’intervalles de variation pour les hyperparamètres et d’une procédure d’exploration de cet espace.
Exploration de l’espace des hyperparamètres suivant la procédure choisie. Pour chaque valeur considérée des hyperparamètres, évaluation des modèles résultants par application de la validation croisée.
Comparaison des résultats de validation croisée, sélection des valeurs des hyperparamètres qui mènent aux meilleures performances (à l’erreur de généralisation estimée la plus faible).
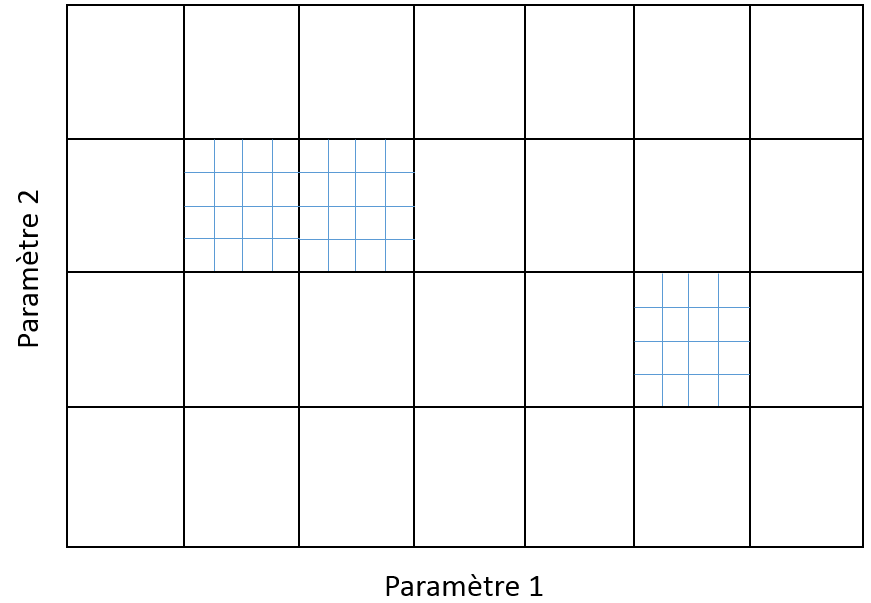
Une procédure d’exploration simple est la recherche dans une grille (grid search) : si on dispose de \(m\) hyperparamètres, on définit pour chaque hyperparamètre un intervalle et un pas de variation, on obtient ainsi une grille (de dimension \(m\)) dont les nœuds correspondent aux valeurs à tester. Pour chaque nœud de la grille, la validation croisée est appliquée afin d’obtenir une estimation de l’erreur de généralisation des modèles correspondants. Bien entendu, tous les nœuds de la grille peuvent être explorés en parallèle.
Avec une grille trop fine, le nombre de modèles à explorer peut être excessif. Avec une grille trop grossière, l’exploration de l’espace des paramètres peut être trop grossière et ne pas permettre de trouver les meilleurs modèles. Une solution souvent adoptée est celle d’une grille qui a plusieurs niveaux de « finesse », comme dans la figure suivante. Dans un tel cas, la recherche est hiérarchique : sont d’abord explorées les valeurs correspondant à la grille grossière, ensuite des grilles plus fines sont définies dans les intervalles de la grille grossière où les meilleurs résultats ont été trouvés, et sont explorées enfin les valeurs issues de ces grilles plus fines. Le procédé peut être répété à plusieurs niveaux. A chaque niveau, les nœuds de la grille peuvent être explorés en parallèle.
Fig. 83 Grille bidimensionnelle à deux niveaux de granularité¶
Il est important de noter que pour des méthodes d’apprentissage spécifiques il est parfois possible de définir des procédures d’exploration plus efficaces.
Enfin, pour estimer le risque espéré (l’erreur de généralisation) du modèle retenu in fine il faut employer des données qui n’ont été utilisées ni pour l’apprentissage du modèle (pour l’algorithme d’optimisation des paramètres), ni pour la recherche des meilleures valeurs des hyperparamètres !
Spark ML est une interface de programmation (API) conçue pour permettre de définir et évaluer des pipelines (chaînes de traitement) de plusieurs outils de modélisation et apprentissage statistique. Plusieurs notions sont importantes dans ce contexte :
Dataset : structure de données distribuée représentée par une DataFrame qui généralise la notion de RDD à des relations définies par des « colonnes » nommées (attributs) pouvant être de types différents.
Transformer : méthode qui transforme une DataFrame en une autre DataFrame. Par exemple, une méthode de réduction de dimension qui modifie la représentation des observations, ou un modèle décisionnel qui fait des prédictions à partir de données d’entrée.
Estimator : algorithme qui produit un Transformer à partir d’une DataFrame. Par exemple, un modèle décisionnel est obtenu par apprentissage sur les données représentées par une DataFrame.
Pipeline : chaîne de traitement composée d’une succession de Estimator et Transformer.
Param : outil de spécification de paramètres, ou de la recherche de paramètres, pour un ou plusieurs Estimator et Transformer.
Spark ML permet l’utilisation de la recherche en grille (voir ParamGridBuilder) et la sélection de modèle (ou de « procédure » de modélisation) par validation croisée (k-fold, voir CrossValidator).
Duc Dung Nguyen, Tu Bao Ho. An efficient method for simplifying support vector machines. Proc. 22nd Intl. Conf. on Machine Learning (ICML’05). ACM, New York, NY, USA, 617-624.