La classification spectrale vise à obtenir un partitionnement des données (ou observations) en groupes à partir d’une représentation de ces données sous la forme d’un graphe de similarités. Dans un tel graphe, chaque sommet correspond à une donnée (ou observation) et chaque arête qui relie deux observations est pondérée par la similarité entre ces observations.
Plus précisément, soit un ensemble \(\mathcal{E} = \{\mathbf{x}_i, 1 \leq i \leq N\}\) de \(N\) données décrites par une matrice de similarités \(\mathbf{S}\) telle que l’élément \(s_{ij} \geq 0\) soit la similarité entre \(\mathbf{x}_i\) et \(\mathbf{x}_j\).
L’objectif de la classification automatique est de répartir les \(N\) données de \(\mathcal{E}\) en \(k\) groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) (inconnus a priori) tels que les similarités soient fortes intra-groupe et faibles entre les groupes.
La méthode de classification spectrale procède en plusieurs étapes :
A partir de \(\mathbf{S}\) est obtenu un graphe de similarités. Plusieurs méthodes alternatives de construction d’un tel graphe sont examinées dans la suite.
A l’aide du graphe, une représentation vectorielle « convenable » (du point de vue de la classification automatique) des données est obtenue.
Un algorithme de classification de base (comme K-means) est enfin appliqué pour obtenir les \(k\) groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\).
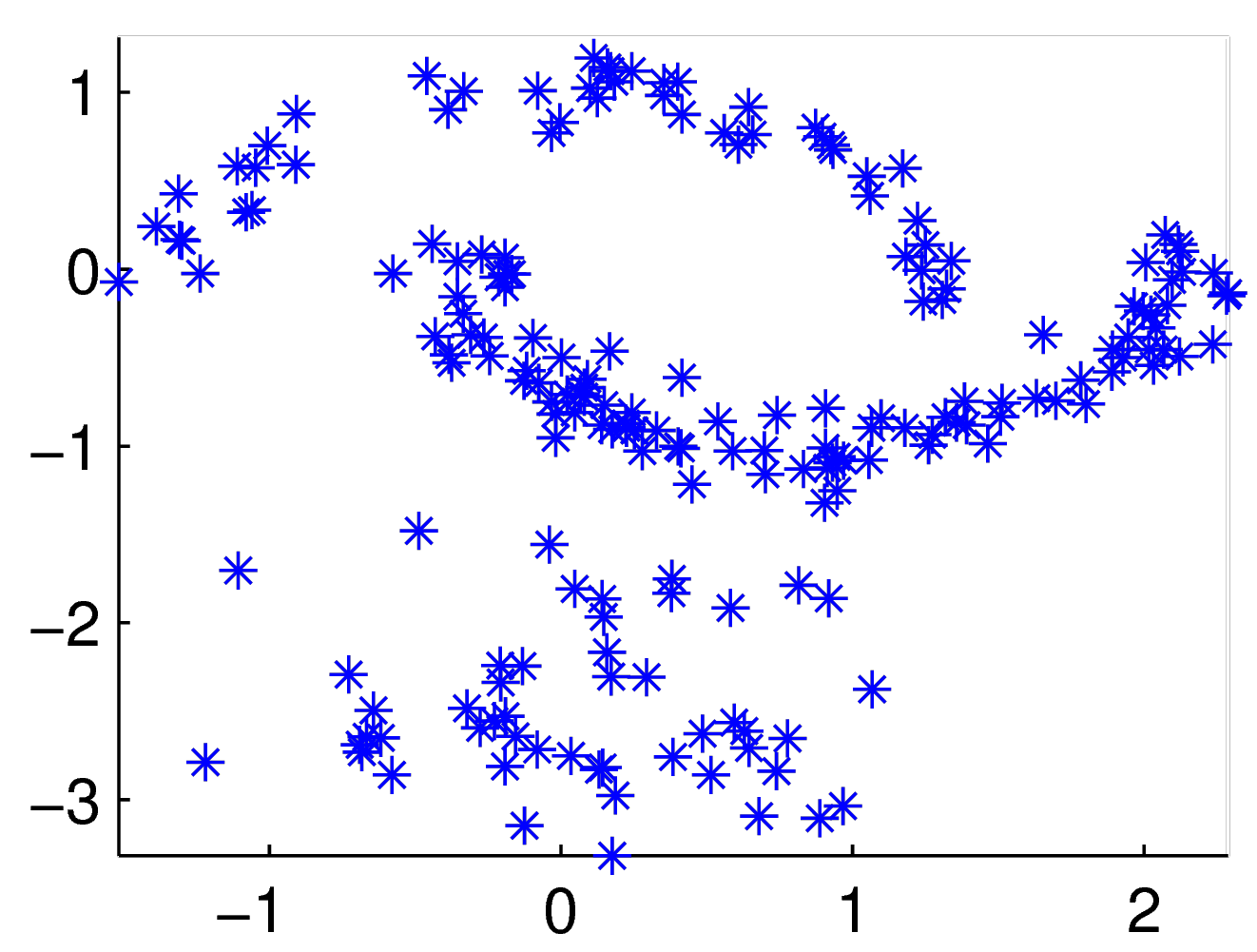
Afin d’illustrer le fonctionnement de la classification spectrale, nous nous servirons dans la suite de l’exemple donné dans [Lux07]. Les données sont des vecteurs dans l’espace bidimensionnel, illustrés dans la figure suivante par des étoiles bleues.
Données pour l’exemple de classification spectrale (issu de [Lux07])¶
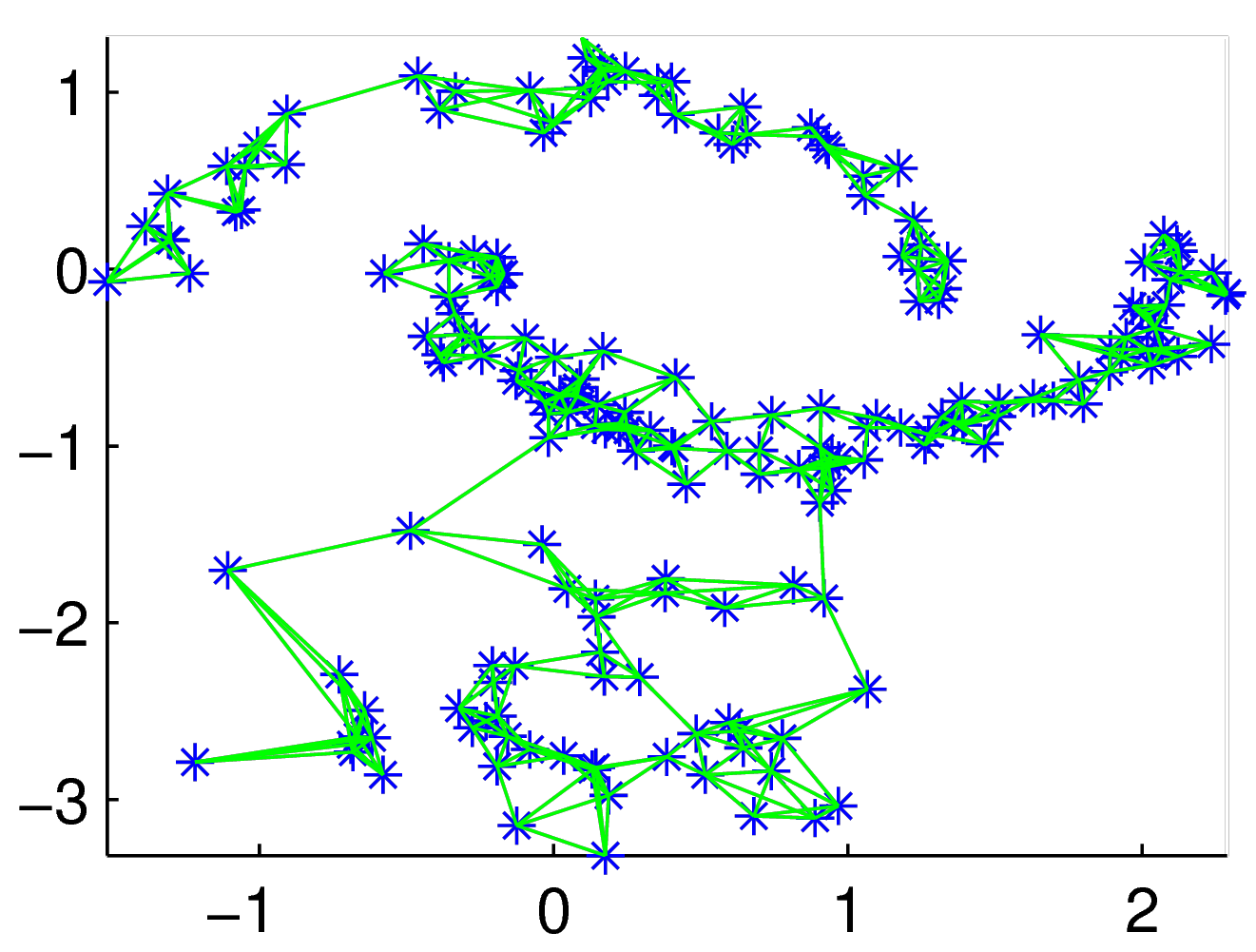
A partir de ces données vectorielles, une matrice de similarités est obtenue et ensuite un graphe de similarités est déduit. Nous examinons plus loin les méthodes permettant de calculer des similarités à partir des distances et d’obtenir un graphe à partir de la matrice de similarités. Dans l’exemple considéré nous pouvons illustrer un graphe de similarités par la figure suivante.
Graphe de similarités pour l’exemple (issu de [Lux07])¶
Avant d’aller plus loin, il est nécessaire d’introduire (ou rappeler) quelques notions concernant les graphes.
Un graphe non orienté est un couple \(G = (V,E)\), où \(V = \{v_1, \ldots, v_N\}\) est l’ensemble des (\(N\)) sommets et \(E\) l’ensemble des arêtes. Voir l’exemple de graphe de la figure précédente, où les sommets sont les données (vecteurs de \(\mathbb{R}^2\) dans ce cas).
Les arêtes peuvent être pondérées : soit \(w_{ij} \geq 0\) le poids de l’arête qui lie les sommets \(v_i\) et \(v_j\), alors \(w_{ij} = 0\) si et seulement si (ssi) il n’y a pas d’arête entre les sommets \(v_i\) et \(v_j\). La matrice des poids est \(\mathbf{W}\) d’élément générique \(w_{ij}\).
Le degré du sommet \(v_i\) est la somme des poids des arêtes qui relient ce sommet à d’autres, \(d_i = \sum_{j=1}^N w_{ij}\). On peut considérer une matrice diagonale des degrés \(\mathbf{D}\) qui a \(\{d_1, \ldots, d_N\}\) sur la diagonale principale.
Une chaîne ou un chemin\(\mu(v_i,v_j)\) entre les sommets \(v_i\) et \(v_j\) est une suite d’arêtes de \(E\) permettant de relier les deux sommets.
Un sous-ensemble \(C \subset V\) forme une composante connexe du graphe \(G = (V,E)\) si
quels que soient deux sommets de \(C\), il existe une chaîne de \(G\) les reliant (\(\forall v_i,v_j \in C\), \(\exists \mu(v_i,v_j)\)) et
pour tout sommet \(v_i\) de \(C\) et toute chaîne avec des arêtes de \(E\) et partant de \(v_i\), le sommet terminal de la chaîne est également dans \(C\) (\(\forall v_i \in C, \forall \mu(v_i,v_p)\), \(v_p \in C\)).
Le vecteur indicateur d’un sous-ensemble \(C\) de \(V\) est le vecteur \(\mathbf{c}\) à \(N\) composantes binaires tel que \(c_i = 1\) si le sommet \(v_i \in C\) et \(c_i = 0\) sinon. La même définition s’applique lorsque \(C\) est une composante connexe.
Pour un graphe non orienté à arêtes pondérées, la matrice laplacienne (non normalisée) est définie par \(\mathbf{L} = \mathbf{D} - \mathbf{W}\). La matrice laplacienne possède plusieurs propriétés importantes :
pour tout vecteur \(\mathbf{x} \in \mathbb{R}^N\), nous avons \(\mathbf{x}^T \mathbf{L} \mathbf{x} = \frac{1}{2} \sum_{i,j=1}^N w_{ij} (x_i - x_j)^2\) ;
la matrice laplacienne \(\mathbf{L}\) est symétrique (\(\mathbf{L} = \mathbf{L}^T\)) et semi-définie positive (\(\forall \mathbf{x} \in \mathbb{R}^N\), \(\mathbf{x}^T \mathbf{L} \mathbf{x} \geq 0\));
la matrice laplacienne \(\mathbf{L}\) a \(N\) valeurs propres réelles \(0 = \lambda_1 \leq \lambda_2 \leq \ldots \leq \lambda_N\) ;
la valeur propre la plus faible de \(\mathbf{L}\) est 0, le vecteur propre correspondant étant le vecteur constant \(\mathbf{1}\) (toutes ses composantes sont égales à \(1\)) ; cela s’explique facilement par la définition même des degrés des sommets ;
la multiplicité de la valeur propre 0 de \(\mathbf{L}\) est égale au nombre de composantes connexes de \(G\) ;
les vecteurs indicateurs de ces composantes connexes sont des vecteurs propres de \(\mathbf{L}\) pour la valeur propre 0.
Plusieurs méthodes peuvent être employées pour normaliser la matrice laplacienne d’un graphe, notamment :
Nous pouvons maintenant détailler les étapes 2 et 3 de l’algorithme de classification spectrale :
A partir de \(\mathbf{S}\) est produit un graphe de similarités \(G\), avec les pondérations \(w_{ij} = s_{ij}\).
A l’aide du graphe, une représentation vectorielle « convenable » (du point de vue de la classification automatique) des données est obtenue par la méthode suivante :
Calculer la matrice laplacienne \(\mathbf{L}\) ;
Calculer les \(k\) vecteurs propres \(\mathbf{u}_1, \ldots, \mathbf{u}_k\) associés aux \(k\) plus petites valeurs propres de \(\mathbf{L} \mathbf{u} = \lambda \mathbf{D} \mathbf{u}\) ;
Soit \(\mathbf{U}_k\) la matrice dont les colonnes sont les vecteurs \(\mathbf{u}_1, \ldots, \mathbf{u}_k\) ;
Soit \(\mathbf{y}_i \in \mathbb{R}^k, 1 \leq i \leq N\), les \(N\) lignes de la matrice \(\mathbf{U}_k\), chaque donnée (observation) \(i\) est représentée par le vecteur \(\mathbf{y}_i\).
Un algorithme de classification de base (comme K-means) est enfin appliqué pour obtenir les \(k\) groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) :
Appliquer K-means aux \(N\) vecteurs \(\mathbf{y}_i\) pour obtenir les groupes \(\mathcal{C}_1, \ldots, \mathcal{C}_k\) ;
Cet algorithme peut être justifié de plusieurs façons différentes. Nous mentionnons dans la suite deux des justifications les plus courantes, sans toutefois détailler complètement les formulations mathématiques correspondantes (qui peuvent être consultées par ex. dans [Lux07] et ses références).
Considérons un graphe non orienté à arêtes pondérées \(G = (V, E)\), que nous souhaitons découper en \(k\) groupes (sous-ensembles) disjoints \(V_1, \ldots, V_k\) de sommets.
La coupe minimum est obtenue en choisissant les \(k\) groupes de sommets de façon à minimiser la somme des poids des arêtes qui relient des sommets appartenant à des groupes différents. Plus précisément, est minimisée la quantité \(\sum_{s=1}^k \omega(V_s, V-V_s)\), où \(\omega(V_s, V_t) = \sum_{v_i \in V_s, v_j \in V_t} w_{ij}\) et \(V-V_s\) est l’ensemble de sommets de \(V\) qui ne sont pas dans \(V_s\). Malheureusement, bien souvent la coupe minimum sépare le graphe en un nombre limité \(l \ll k\) de grands sous-graphes plus quelques sommets isolés, solution jugée en général peu satisfaisante.
Avec la coupe normalisée (normalized cut, Ncut), les sous-ensembles de sommets sont plus équilibrés. La coupe normalisée est obtenue en minimisant \(\sum_{s=1}^k \frac{\omega(V_s, V-V_s)}{\textrm{vol}(V_s)}\), où le « volume » d’une composante \(V_s\) est la somme des degrés des sommets de cette composante, \(\textrm{vol}(V_s) = \sum_{v_i \in V_s} d_i\). Malheureusement, si la solution obtenue est en général plus satisfaisante, ce problème de minimisation est NP-difficile !
La classification spectrale reformule le problème de minimisation en utilisant un vecteur indicateur pour chaque sous-ensemble de sommets, avec une « relaxation » continue du problème discret : les composantes du vecteur indicateur ne sont pas binaires mais ont des valeurs dans l’intervalle \([0, 1]\). Considérons la matrice \(\mathbf{U}_k\) dont les colonnes sont les vecteurs caractéristiques \(\mathbf{u}_1, \ldots, \mathbf{u}_k\) des \(k\) groupes recherchés et chacune des \(N\) lignes correspond à une des observations.
Avec des groupes disjoints, chacune des observations appartient à un seul groupe et donc le vecteur \(\mathbf{y}_i\) qui représente l’observation \(i\) a une seule composante égale à 1 (la composante \(y_{ij}\) si l’observation \(i\) fait partie du groupe \(j\)), alors que les autres sont égales à 0. Avec la relaxation continue, ces composantes du vecteur ont toutes des valeurs dans l’intervalle \([0, 1]\) ; on peut les voir comme des degrés d’appartenance de cette observation aux différents groupes.
Avec cette relaxation continue, la coupe normalisée est ainsi traduite en un problème de minimisation sous contraintes d’égalité. Les contraintes d’égalité consistent à imposer, pour toute ligne de \(\mathbf{U}_k\), que la somme des composantes soit égale à 1 : \(\forall i \in \{1, \ldots, N\}, \sum_{j=1}^k y_{ij} = 1\).
Pour résoudre ce nouveau problème de minimisation, il faut trouver les \(k\) plus petites valeurs propres de \(\mathbf{L} \mathbf{u} = \lambda \mathbf{D} \mathbf{u}\) et les vecteurs propres associés (qui seront les colonnes de \(\mathbf{U}_k\)). En l’absence de sommets isolés, \(\mathbf{L} \mathbf{u} = \lambda \mathbf{D} \mathbf{u}\) devient \(\mathbf{D}^{-1} \mathbf{L} \mathbf{u} = \lambda \mathbf{u}\), ou encore \(\mathbf{L}_{rw} \mathbf{u} = \lambda \mathbf{u}\).
Pour revenir à des vecteurs caractéristiques binaires, dans la dernière étape de la classification spectrale un algorithme de classification automatique de base (comme K-means) est appliqué aux lignes de \(\mathbf{U}_k\) et ainsi chaque donnée est affectée à un groupe (chaque sommet est affecté à une composante du graphe).
On arrive au même problème de coupe normalisée en cherchant à identifier des composantes du graphe telles qu’une marche aléatoire a peu d’opportunités de passer d’une composante à une autre. Une marche aléatoire sur un graphe est une succession de sauts aléatoires d’un sommet du graphe à un autre sommet, suivant des probabilités de transition. La probabilité de passer du sommet \(i\) au sommet \(j\) (probabilité de transition \(i \rightarrow j\)) est le rapport entre le poids de l’arête (ici, similarité entre les sommets \(i\) et \(j\)) et la somme des poids des arêtes partant du sommet \(i\) (le degré de ce sommet) : \(p_{ij} = w_{ij} / d_i\).
Avec ces probabilités de transition, la matrice de transition est \(\mathbf{P} = \mathbf{D}^{-1} \mathbf{W}\), c’est à dire \(\mathbf{P} = \mathbf{I}_N - \mathbf{L}_{rw}\). Alors, \(\lambda\) est une valeur propre de \(\mathbf{L}_{rw}\) avec comme vecteur propre \(\mathbf{u}\) si et seulement si \(1-\lambda\) est une valeur propre de \(\mathbf{P}\) avec le même vecteur propre \(\mathbf{u}\).
On peut montrer que trouver \(V_s \subset V\) pour minimiser la probabilité de passage de \(V_s\) à \(V - V_s\) et de \(V - V_s\) à \(V_s\) est équivalent au problème de la coupe normalisée. La classification spectrale fournit une solution en général approximative au problème de la coupe normalisée et donc également à l’identification de composantes d’un graphe telles qu’une marche aléatoire a peu d’opportunités de passer d’une composante à une autre.
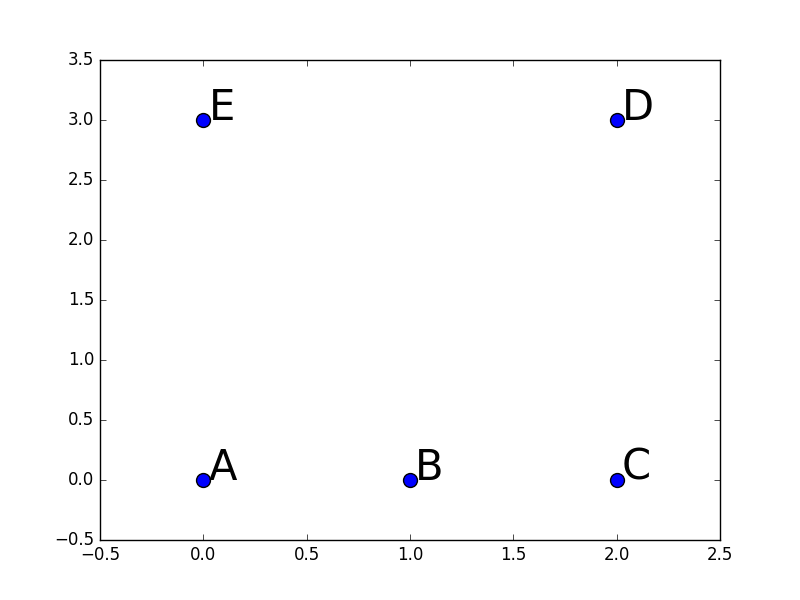
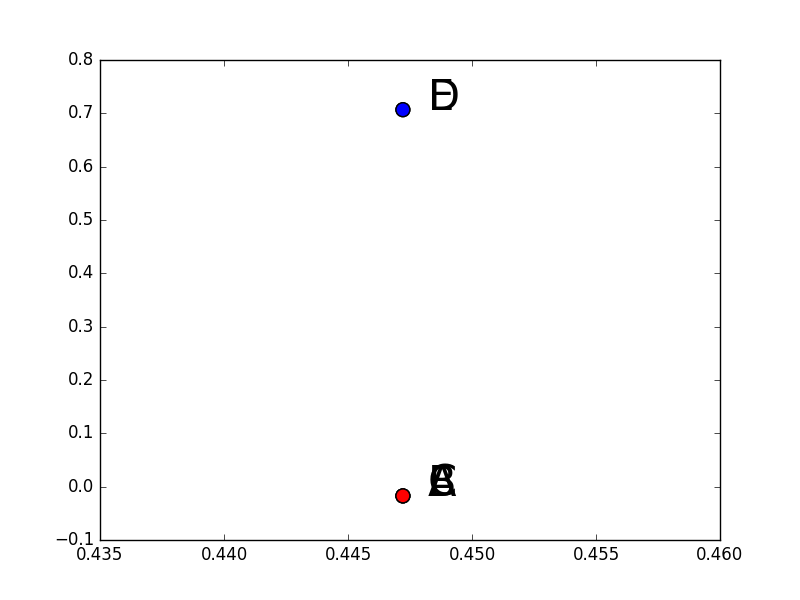
Deux exemples numériques liés entre eux peuvent aider à mieux comprendre comment est obtenue la représentation vectorielle « convenable » (du point de vue de la classification automatique) des données. Considérons les 5 données (observations) bidimensionnelles qui sont les lignes de la matrice suivante :
Ces données sont représentées aussi dans la figure ci-dessous :
Données pour l’exemple numérique 1 de classification spectrale¶
En faisant attention au fait que l’échelle de l’abscisse n’est pas tout à fait la même que celle de l’ordonnée, nous pouvons constater visuellement que le choix de 2 groupes ({A, B, C} et {D, E}) serait naturel dans ce cas, suivi par le choix de 3 groupes ({A, B, C}, {D}, {E}).
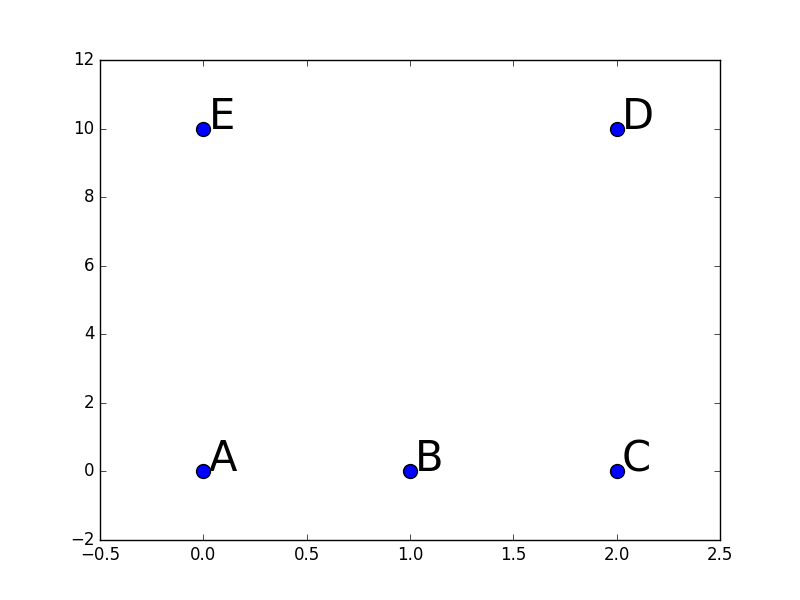
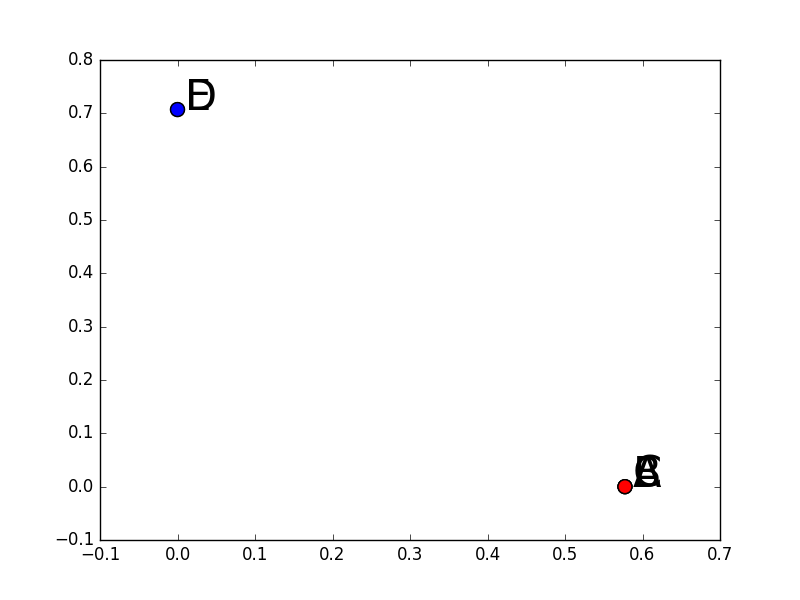
Le deuxième exemple est issu du premier en écartant beaucoup plus les observations D et E de A, B et C, au point où cela peut produire la déconnexion entre deux composantes du graphe :
La figure suivante illustre ce deuxième exemple (attention, l’échelle verticale est très différente) :
Données pour l’exemple numérique 2 de classification spectrale¶
Dans ce cas, par la visualisation on constate que le choix naturel est celui de 2 groupes : {A, B, C} et {D, E}.
Nous utilisons la distance euclidienne classique pour mesurer la distance entre les observations. La matrice des similarités est obtenue à partir du noyau gaussien de variance 1 appliqué à la distance euclidienne. Les matrices de similarités résultantes (valeurs arrondies à 6 décimales) sont alors :
On observe que le graphe associé au premier exemple peut être considéré complètement connecté car toutes les similarités sont strictement supérieures à 0. En revanche, pour le second exemple, avec la précision de représentation considérée, les similarités entre les observations de l’ensemble {A, B, C} et celles de l’ensemble {D, E} sont nulles (valeurs représentées en gras dans la matrice), le graphe associé possède donc deux composantes connexes, {A, B, C} et {D, E}.
Les matrices laplaciennes normalisées asymétriques \(\mathbf{L}_{rw} = \mathbf{D}^{-1} \mathbf{L} \left(= \mathbf{I}_N - \mathbf{D}^{-1} \mathbf{W}\right)\) résultantes (valeurs arrondies à 6 décimales) sont :
Pour le premier exemple, les valeurs propres de \(\mathbf{L}_{rw}\) en ordre croissant sont 0.0, 0.0094, 1.0474, 1.9523 et 1.9907. Pour obtenir une classification en 2 groupes nous nous intéressons aux deux plus petites valeurs propres de \(\mathbf{L}_{rw}\), qui sont ici 0.0 et 0.0094. Les colonnes de la matrice \(\mathbf{U}_2\) suivante sont les 2 vecteurs propres correspondants :
Les nouvelles représentations des données pour l’exemple 1 sont alors les lignes de la matrice \(\mathbf{U}_2\). L’illustration suivante montre que les projections sur ces 2 vecteurs propres des observations A, B et C sont confondues (au niveau de précision numérique choisi) et différentes des projections de D et E qui sont également confondues :
Nouvelles représentations des données pour l’exemple numérique 1¶
Nous remarquerons que le vecteur propre associé à la valeur propre 0 a toutes les composantes identiques et ne présente donc aucune utilité pour distinguer entre les (projections des) observations.
Le choix de trois groupes aurait été bien moins naturel car la troisième valeur propre la plus petite est 1.0474, nettement supérieure à la deuxième qui est 0.0094.
Pour le deuxième exemple, les valeurs propres de \(\mathbf{L}_{rw}\) en ordre croissant sont 0.0, 0.0, 1.0474, 1.9525 et 2.0000. La valeur propre 0 est donc de multiplicité 2, ce qui indique qu’il y a bien 2 composantes connexes dans le graphe. La matrice \(\mathbf{U}_2\) dont les colonnes sont les 2 vecteurs propres correspondant à la même valeur propre 0 (de multiplicité 2) est :
Il faut noter que le choix exact des deux vecteurs propres est arbitraire dans le sous-espace propre à 2 dimensions correspondant à la valeur propre 0 (la dimension de l’espace propre est égale à la multiplicité de la valeur propre). Les seules contraintes sont l’orthogonalité de ces deux vecteurs propres et le fait que chacun a une norme égale à 1.
Les nouvelles représentations des données pour l’exemple 2 sont alors les lignes de la matrice \(\mathbf{U}_2\). L’illustration suivante montre que pour ce deuxième exemple aussi les projections sur ces 2 vecteurs propres des observations A, B et C sont confondues et différentes des projections de D et E qui sont également confondues :
Nouvelles représentations des données pour l’exemple numérique 2¶
Pour ces deux exemples, l’application d’un algorithme de classification comme k-means sur les nouvelles représentations des données afin d’obtenir 2 groupes devrait donner bien plus souvent les bons résultats.
Examinons maintenant la première étape de l’algorithme de classification spectrale, c’est à dire le passage d’une matrice de similarités \(\mathbf{S}\) à un graphe de similarités \(G\).
Une étape préalable souvent nécessaire est de définir une mesure de similarité adaptée aux données de \(\mathcal{E} = \{\mathbf{x}_i, 1 \leq i \leq N\}\). En effet, les données peuvent être de nature diverse : vecteurs (cas classique de données décrites par un ensemble de variables numériques), ensembles (par ex. l’ensemble des mots d’un texte), arbres (par ex. arbres d’analyse grammaticale de phrases), etc. La compréhension des données (quelle est leur nature, quelles données sont similaires et lesquelles ne le sont pas) est très utile pour choisir une « bonne » mesure de similarité. Des mesures de similarités sont souvent définies à partir de fonctions-noyau, comme la loi normale multidimensionnelle pour des données vectorielles. De manière générale, ce qui compte le plus est le comportement de la mesure de similarité pour des données très similaires car les données très dissimilaires ne se retrouveront pas dans le même groupe.
Lorsqu’on dispose d’une métrique (mesure de distance) considérée adéquate pour les données, il est possible d’employer la loi normale pour définir les similarités : \(s_{ij} = \exp \left(- \frac{d(\mathbf{x}_i, \mathbf{x}_j)^2}{2 \sigma^2}\right)\), où \(d(\mathbf{x}_i, \mathbf{x}_j)\) est la distance entre les données vectorielles \(\mathbf{x}_i\) et \(\mathbf{x}_j\). Le choix de l’écart-type \(\sigma\) est important car, la loi normale étant à décroissance rapide, une valeur trop faible produira trop de valeurs nulles pour les \(s_{ij}\), donc nécessairement un graphe de similarités trop peu connecté. L’écart-type \(\sigma\) est en général choisi proche de la distance la plus grande entre deux données voisines dans l’arbre couvrant minimal des données de \(\mathcal{E}\).
Pour ce qui concerne le passage d’une matrice de similarités \(\mathbf{S}\) à un graphe de similarités \(G\), plusieurs solutions sont possibles :
Une première solution est de considérer un graphe complet (où tous les sommets sont connectés) et d’affecter directement les valeurs de similarités issues de la matrice de similarités comme poids des arêtes du graphe, \(w_{ij} = s_{ij}\). Cela revient à compter exclusivement sur la mesure de similarité pour partitionner le graphe (les observations). Il faut également noter que plus le graphe est connecté, plus le coût d’exécution des étapes ultérieures de l’algorithme de classification spectrale est élevé.
Une autre solution est de limiter la connectivité dans le graphe au \(\epsilon\)-voisinage des sommets : deux sommets \(v_i, v_j \in V\) sont connectés si leur similarité est supérieurs à un seuil, \(s_{ij} \geq \epsilon\). L’utilisation d’un seuil \(\epsilon\) est toutefois inadaptée aux situations où des groupes différents de données ont des densités très différentes : une valeur \(\epsilon\) faible limitera fortement la connectivité à l’intérieur des groupes peu denses, alors qu’une valeur plus élevée risque de connecter plusieurs groupes denses entre eux. Aussi, de nombreux sommets peuvent se retrouver isolés (voir la figure suivante) et le restent lors des étapes suivantes de l’algorithme.
Limiter la connectivité dans le graphe aux \(k\) plus proches voisins (\(k\)ppv, k-nearest neighbors, kNN) des sommets répond à ce problème de présence de groupes de densités très différentes. Deux sommets \(v_i, v_j \in V\) sont ainsi connectés si \(v_i\) fait partie des \(k\)ppv de \(v_j\)ou\(v_j\) fait partie des \(k\)ppv de \(v_i\). Cette solution, conseillée en première approche, est relativement robuste au choix de la valeur de \(k\). En revanche, elle mène souvent à la création d’arêtes entre groupes de densités différentes, comme le montre la figure suivante.
L’utilisation des \(k\) plus proches voisins mutuels (mutual k-nearest neighbors) permet de réduire le nombre d’arêtes entre groupes de densités différentes. Deux sommets \(v_i, v_j \in V\) sont connectés si chacun est dans les \(k\) plus proches voisins de l’autre : \(v_i\) fait partie des \(k\)ppv de \(v_j\)et\(v_j\) fait partie des \(k\)ppv de \(v_i\).
Alternatives pour la construction du graphe (exemple issu de [Lux07])¶
Pour comprendre l’idée directrice du choix des paramètres pour ces solutions alternatives il est important de se rappeler que la multiplicité de la valeur propre 0 de \(\mathbf{L}\) est égale au nombre de composantes connexes de \(G\). Or, l’algorithme de classification spectrale s’intéresse aux \(k\) plus petites valeurs propres de \(\mathbf{L}_{rw}\). En conséquence, le nombre de groupes issu de l’algorithme de classification spectrale ne peut être inférieur au nombre de composantes connexes du graphe de similarités. Mieux vaut donc éviter que le graphe \(G\) possède trop de composantes connexes car cela imposerait un nombre de groupes élevé ; une connectivité plus forte est préférable.
Supposons que le graphe \(G\) est construit à partir des \(\epsilon\)-voisinages et considérons que \(\epsilon\) est un seuil supérieur de distance et non un seuil inférieur de similarité. Pour des données vectorielles \(\mathbf{x}_i \in \mathbb{R}^d\) et la distance euclidienne classique, afin d’obtenir un graphe connecté (non décomposable en plusieurs composantes connexes) il faut choisir \(\epsilon \sim \left(\frac{\log(N)}{N}\right)^d\). Pour une métrique quelconque et des données non nécessairement vectorielles, \(\epsilon\) doit être proche de la distance la plus grande entre deux données voisines dans l’arbre couvrant minimal des données de \(\mathcal{E}\).
Si le graphe est construit à partir des \(k\) plus proches voisins, le choix de \(k\) doit respecter \(k \sim \log(N) + 1\). Lorsque la construction de \(G\) est faite grâce aux \(k\) plus proches voisins mutuels, la valeur de \(k\) devrait être plus élevée que pour les \(k\) plus proches voisins simples car pour une même valeur de \(k\) ces graphes basés sur les voisins mutuels ont moins d’arêtes que les graphes basés sur les voisins simples.
Enfin, si le graphe \(G\) est construit directement avec \(w_{ij} = s_{ij}\), le réglage doit porter plutôt sur la fonction de similarité, comme nous l’avons vu plus haut pour le passage d’une métrique à une mesure de similarité à l’aide de la loi normale.
Le nombre de groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) est noté par \(k\), à ne pas confondre avec le nombre de voisins employés dans la construction du graphe (notation traditionnelle). Pour la classification spectrale, le choix du nombre de groupes peut se faire à l’aide d’un critère spécifique, le premier « saut » dans la croissance des valeurs propres (en partant de la plus basse), appelé en anglais eigengap.
Considérons les valeurs propres de la matrice laplacienne \(\mathbf{L}_{rw}\) triées en ordre croissant, \(0 = \lambda_1 \leq \lambda_2 \leq \ldots\) La valeur de \(k\) (nombre de groupes) est choisie telle que dans cette liste triée on trouve un saut entre \(\lambda_k\) et \(\lambda_{k+1}\).
L’exemple suivant, issu de [Lux07], montre deux cas très simples dans lesquels les données sont unidimensionnelles. Les données sont représentées sur l’abscisse, alors que l’ordonnée indique le nombre de données de même abscisse. Dans le premier cas (à gauche) quatre groupes sont clairement identifiables et le graphique correspondant des valeurs propres montre un premier saut entre \(\lambda_4\) et \(\lambda_5\), puis un second entre \(\lambda_8\) et \(\lambda_9\) ; la valeur \(k=4\) sera ainsi retenue. Dans le second cas (à droite), les groupes sont nettement moins bien séparés, en revanche un premier saut reste bien visible entre \(\lambda_4\) et \(\lambda_5\) ; la valeur \(k=4\) sera choisie ici encore.
Illustration du saut dans les valeurs propres (eigengap, exemple issu de [Lux07])¶
Comment obtenir un graphe de similarités¶
Examinons maintenant la première étape de l’algorithme de classification spectrale, c’est à dire le passage d’une matrice de similarités \(\mathbf{S}\) à un graphe de similarités \(G\).
Une étape préalable souvent nécessaire est de définir une mesure de similarité adaptée aux données de \(\mathcal{E} = \{\mathbf{x}_i, 1 \leq i \leq N\}\). En effet, les données peuvent être de nature diverse : vecteurs (cas classique de données décrites par un ensemble de variables numériques), ensembles (par ex. l’ensemble des mots d’un texte), arbres (par ex. arbres d’analyse grammaticale de phrases), etc. La compréhension des données (quelle est leur nature, quelles données sont similaires et lesquelles ne le sont pas) est très utile pour choisir une « bonne » mesure de similarité. Des mesures de similarités sont souvent définies à partir de fonctions-noyau, comme la loi normale multidimensionnelle pour des données vectorielles. De manière générale, ce qui compte le plus est le comportement de la mesure de similarité pour des données très similaires car les données très dissimilaires ne se retrouveront pas dans le même groupe.
Lorsqu’on dispose d’une métrique (mesure de distance) considérée adéquate pour les données, il est possible d’employer la loi normale pour définir les similarités : \(s_{ij} = \exp \left(- \frac{d(\mathbf{x}_i, \mathbf{x}_j)^2}{2 \sigma^2}\right)\), où \(d(\mathbf{x}_i, \mathbf{x}_j)\) est la distance entre les données vectorielles \(\mathbf{x}_i\) et \(\mathbf{x}_j\). Le choix de l’écart-type \(\sigma\) est important car, la loi normale étant à décroissance rapide, une valeur trop faible produira trop de valeurs nulles pour les \(s_{ij}\), donc nécessairement un graphe de similarités trop peu connecté. L’écart-type \(\sigma\) est en général choisi proche de la distance la plus grande entre deux données voisines dans l’arbre couvrant minimal des données de \(\mathcal{E}\).
Pour ce qui concerne le passage d’une matrice de similarités \(\mathbf{S}\) à un graphe de similarités \(G\), plusieurs solutions sont possibles :
Une première solution est de considérer un graphe complet (où tous les sommets sont connectés) et d’affecter directement les valeurs de similarités issues de la matrice de similarités comme poids des arêtes du graphe, \(w_{ij} = s_{ij}\). Cela revient à compter exclusivement sur la mesure de similarité pour partitionner le graphe (les observations). Il faut également noter que plus le graphe est connecté, plus le coût d’exécution des étapes ultérieures de l’algorithme de classification spectrale est élevé.
Une autre solution est de limiter la connectivité dans le graphe au \(\epsilon\)-voisinage des sommets : deux sommets \(v_i, v_j \in V\) sont connectés si leur similarité est supérieurs à un seuil, \(s_{ij} \geq \epsilon\). L’utilisation d’un seuil \(\epsilon\) est toutefois inadaptée aux situations où des groupes différents de données ont des densités très différentes : une valeur \(\epsilon\) faible limitera fortement la connectivité à l’intérieur des groupes peu denses, alors qu’une valeur plus élevée risque de connecter plusieurs groupes denses entre eux. Aussi, de nombreux sommets peuvent se retrouver isolés (voir la figure suivante) et le restent lors des étapes suivantes de l’algorithme.
Limiter la connectivité dans le graphe aux \(k\) plus proches voisins (\(k\)ppv, k-nearest neighbors, kNN) des sommets répond à ce problème de présence de groupes de densités très différentes. Deux sommets \(v_i, v_j \in V\) sont ainsi connectés si \(v_i\) fait partie des \(k\)ppv de \(v_j\) ou \(v_j\) fait partie des \(k\)ppv de \(v_i\). Cette solution, conseillée en première approche, est relativement robuste au choix de la valeur de \(k\). En revanche, elle mène souvent à la création d’arêtes entre groupes de densités différentes, comme le montre la figure suivante.
L’utilisation des \(k\) plus proches voisins mutuels (mutual k-nearest neighbors) permet de réduire le nombre d’arêtes entre groupes de densités différentes. Deux sommets \(v_i, v_j \in V\) sont connectés si chacun est dans les \(k\) plus proches voisins de l’autre : \(v_i\) fait partie des \(k\)ppv de \(v_j\) et \(v_j\) fait partie des \(k\)ppv de \(v_i\).
Pour comprendre l’idée directrice du choix des paramètres pour ces solutions alternatives il est important de se rappeler que la multiplicité de la valeur propre 0 de \(\mathbf{L}\) est égale au nombre de composantes connexes de \(G\). Or, l’algorithme de classification spectrale s’intéresse aux \(k\) plus petites valeurs propres de \(\mathbf{L}_{rw}\). En conséquence, le nombre de groupes issu de l’algorithme de classification spectrale ne peut être inférieur au nombre de composantes connexes du graphe de similarités. Mieux vaut donc éviter que le graphe \(G\) possède trop de composantes connexes car cela imposerait un nombre de groupes élevé ; une connectivité plus forte est préférable.
Supposons que le graphe \(G\) est construit à partir des \(\epsilon\)-voisinages et considérons que \(\epsilon\) est un seuil supérieur de distance et non un seuil inférieur de similarité. Pour des données vectorielles \(\mathbf{x}_i \in \mathbb{R}^d\) et la distance euclidienne classique, afin d’obtenir un graphe connecté (non décomposable en plusieurs composantes connexes) il faut choisir \(\epsilon \sim \left(\frac{\log(N)}{N}\right)^d\). Pour une métrique quelconque et des données non nécessairement vectorielles, \(\epsilon\) doit être proche de la distance la plus grande entre deux données voisines dans l’arbre couvrant minimal des données de \(\mathcal{E}\).
Si le graphe est construit à partir des \(k\) plus proches voisins, le choix de \(k\) doit respecter \(k \sim \log(N) + 1\). Lorsque la construction de \(G\) est faite grâce aux \(k\) plus proches voisins mutuels, la valeur de \(k\) devrait être plus élevée que pour les \(k\) plus proches voisins simples car pour une même valeur de \(k\) ces graphes basés sur les voisins mutuels ont moins d’arêtes que les graphes basés sur les voisins simples.
Enfin, si le graphe \(G\) est construit directement avec \(w_{ij} = s_{ij}\), le réglage doit porter plutôt sur la fonction de similarité, comme nous l’avons vu plus haut pour le passage d’une métrique à une mesure de similarité à l’aide de la loi normale.
Le nombre de groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) est noté par \(k\), à ne pas confondre avec le nombre de voisins employés dans la construction du graphe (notation traditionnelle). Pour la classification spectrale, le choix du nombre de groupes peut se faire à l’aide d’un critère spécifique, le premier « saut » dans la croissance des valeurs propres (en partant de la plus basse), appelé en anglais eigengap.
Considérons les valeurs propres de la matrice laplacienne \(\mathbf{L}_{rw}\) triées en ordre croissant, \(0 = \lambda_1 \leq \lambda_2 \leq \ldots\) La valeur de \(k\) (nombre de groupes) est choisie telle que dans cette liste triée on trouve un saut entre \(\lambda_k\) et \(\lambda_{k+1}\).
L’exemple suivant, issu de [Lux07], montre deux cas très simples dans lesquels les données sont unidimensionnelles. Les données sont représentées sur l’abscisse, alors que l’ordonnée indique le nombre de données de même abscisse. Dans le premier cas (à gauche) quatre groupes sont clairement identifiables et le graphique correspondant des valeurs propres montre un premier saut entre \(\lambda_4\) et \(\lambda_5\), puis un second entre \(\lambda_8\) et \(\lambda_9\) ; la valeur \(k=4\) sera ainsi retenue. Dans le second cas (à droite), les groupes sont nettement moins bien séparés, en revanche un premier saut reste bien visible entre \(\lambda_4\) et \(\lambda_5\) ; la valeur \(k=4\) sera choisie ici encore.
von Luxburg, U. A tutorial on spectral clustering. CoRR, abs/0711.0189, 2007.