La classification automatique (cluster analysis ou clustering en anglais) cherche à répartir un ensemble donné de \(N\) observations en groupes (catégories, classes, taxons, clusters) de façon à regrouper les observations similaires et à séparer les observations dissimilaires. Dans la mesure où les notions de similarité et de groupe peuvent être explicitées de multiples façons, de nombreuses méthodes de classification automatique ont été proposées depuis les années 1930. Des synthèses partielles peuvent être trouvées, par exemple, dans [JMF99] ou [BBS14]. Le choix d’une méthode adéquate exige une connaissance des données et de la nature des groupes recherchés.
En général, nous pouvons parler de classification automatique si aucune information n’est disponible concernant l’appartenance de certaines données à certaines classes connues a priori. Par ailleurs, le nombre de groupes recherchés peut être connu a priori ou non.
Une première distinction entre méthodes de classification automatique peut être faite suivant leur objectif. La plupart des méthodes visent à obtenir un partitionnement des données, comme dans l’exemple suivant, plus éventuellement à trouver une donnée « représentative » (« prototype ») par groupe :
Fig. 60 Exemple de partitionnement de données bidimensionnelles en 3 groupes¶
D’autres méthodes cherchent plutôt à obtenir une hiérarchie de regroupements, qui fournit une information plus riche concernant la structure de similarité des données. Noter qu’à partir d’une telle hiérarchie il est facile d’extraire plusieurs partitionnements, à des niveaux de « granularité » différents, comme dans l’exemple suivant :
Fig. 61 Exemple de regroupement hiérarchique qui peut servir à obtenir plusieurs partitionnements¶
Dans les deux cas, la classification automatique permet de
mettre en évidence une structure simple (partitionnement ou hiérarchie de groupes) dans un ensemble de données ;
résumer un ensemble de données par les représentants des groupes (« prototypes »), éventuellement à plusieurs niveaux hiérarchiques.
Une seconde distinction entre méthodes de classification automatique peut être faite suivant la nature des données à regrouper : données numériques, données catégorielles ou données mixtes. Si la plupart des méthodes s’intéressent aux données numériques, certaines peuvent traiter directement des données catégorielles. Toutefois, les données catégorielles sont fréquemment transformées en données numériques (par ex. par une analyse factorielle des correspondances multiples) avant d’être traitées par des méthodes de classification adaptées aux données numériques.
Une autre distinction correspond à la représentation des données. De nombreuses méthodes de classification automatique travaillent sur des représentations vectorielles qui, en plus d’accepter différentes métriques, permettent de définir des centres de gravité de groupes, des densités de probabilité, des intervalles, des sous-espaces, etc. Ces méthodes sont en général basées sur l’utilisation de prototypes et leur complexité est en général de \(O(N)\). Certaines méthodes se satisfont, en revanche, d’une simple structure métrique (distances) sur l’espace des données ; cela leur confère une grande généralité mais également une complexité plus élevée, \(\geq O(N^2)\).
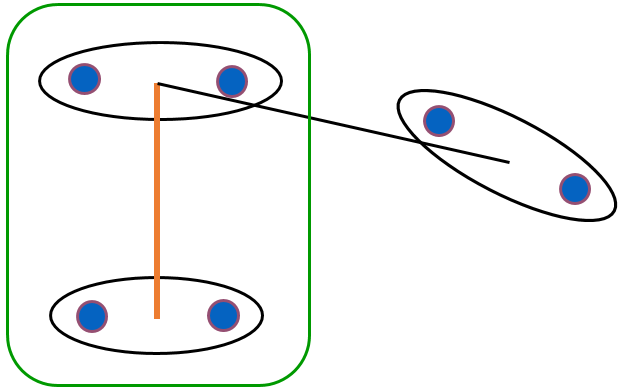
Fig. 62 A quel groupe appartiennent les données entourées ?¶
Une distinction importante concerne la nature des groupes recherchés : sont-il mutuellement exclusifs ou non ? Sont-ils « nets » (crisp, une observation appartient ou n’appartient pas à un groupe) ou « flous » (fuzzy, une observation peut appartenir à différents degrés à plusieurs groupes) ? Si la plupart des méthodes de classification automatique s’intéressent aux partitionnement exclusifs, certaines permettent d’obtenir des groupes non exclusifs. Aussi, des extensions floues existent pour des méthodes de classification automatiques bien connues (par ex. fuzzy c-means pour K-means, voir [JMF99]). Les extensions floues peuvent avoir une convergence plus robuste vers une solution que les méthodes « nettes » de départ. Il est possible de passer des groupes flous à des groupes nets en affectant chaque donnée (ou observation) au groupe auquel elle appartient le plus.
Enfin, une autre distinction importante peut être faite suivant le critère de regroupement. Certaines méthodes cherchent des groupes « compacts » et relativement éloignés entre eux, comme dans cet exemple Fig. 60. D’autres méthodes s’intéressent à des groupes denses (et non nécessairement « compacts ») séparés par des régions moins denses, comme dans l’exemple suivant :
Fig. 63 Exemple de groupes denses séparés par des régions moins denses¶
Il faut noter que ce critère de regroupement n’est pas toujours explicite, pourtant son impact sur les résultats obtenus est majeur.
Considérons un ensemble \(\mathcal{E}\) de \(N\) données décrites par \(p\) variables à valeurs dans \(\mathbb{R}\) et \(d\) une distance sur \(\mathbb{R}^p\). Pour regrouper ces données en \(k\) groupes disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\), inconnus a priori, on s’intéresse souvent à un critère qui correspond à la somme des inerties intra-classe des groupes :
avec \(\mathcal{C} = \{\mathbf{m}_j, 1 \leq j \leq k\}\) l’ensemble des centres de gravité des \(k\) groupes. Pour \(\mathcal{E}\) et \(k\) donnés, plus la valeur de \(\phi_\mathcal{E}(\mathcal{C})\) est faible, plus les groupes sont « compacts » autour de leurs centres et donc meilleure est la qualité du partitionnement obtenu.
La somme des inerties intra-classe peut s’écrire aussi
\[\phi_\mathcal{E}(\mathcal{C}) = \sum_{1 \leq l \leq N} d^2(\mathbf{x}_l,\mathbf{m}_{C(l)})\]
où \({C(l)}\) est l’indice du groupe dont fait partie \(\mathbf{x}_l\). Cela permet de voir que, pour un ensemble \(\mathcal{E}\) donné, les variables dont dépend la valeur du critère sont les positions des centres, \(\{\mathbf{m}_j, 1 \leq j \leq k\}\).
Trouver le minimum global de la fonction (21) est un problème NP-difficile, mais on dispose d’algorithmes de complexité polynomiale dans le nombre de données \(N\) qui produisent une solution en général sous-optimale.
Un tel algorithme est l’algorithme des centres mobiles décrit ci-dessous :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) ;
Sorties : \(k\) groupes (clusters) disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) et ensemble \(\mathcal{C}\) de leurs centres ;
Affectation de chaque donnée au groupe du centre le plus proche ;
Remplacement des centres par les centres de gravité des groupes ;
fintantque
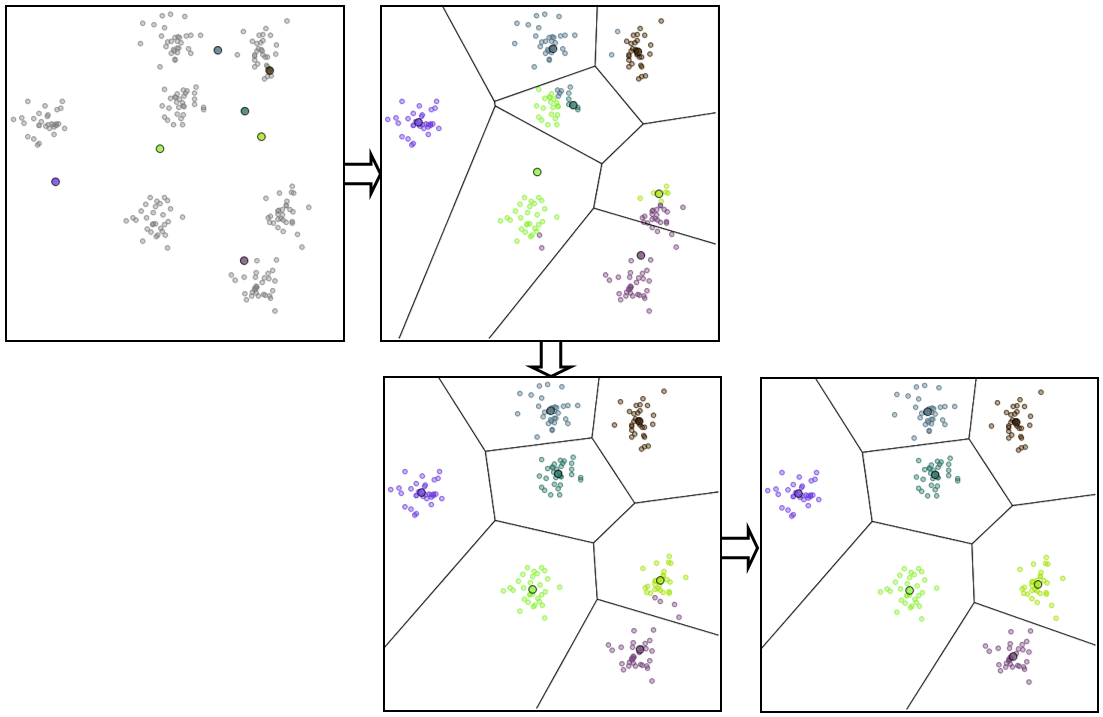
Des étapes successives dans l’application de cet algorithme à des données issues de 7 lois normales bidimensionnelles peuvent être visualisées dans la figure suivante. La distance euclidienne classique est employée et la classification est réalisée avec avec \(k=7\) centres, dont la position initiale (choisie aléatoirement) est indiquée dans la première image.
Fig. 64 Étapes successives dans l’application de K-means¶
Dans cet exemple, la solution obtenue est optimale et est atteinte après seulement 2 itérations.
On peut montrer que la valeur de \(\phi_\mathcal{E}(\mathcal{C})\) diminue lors de chacune des deux étapes du processus itératif (affectation de chaque donnée à un groupe, recalcul des centres) :
Affectation de chaque donnée au groupe du centre le plus proche : \(\mathbf{x}_i\) passe du groupe de centre \(\mathbf{m}_p\) au groupe de centre \(\mathbf{m}_q\) si \(d^2(\mathbf{x}_i,\mathbf{m}_p) > d^2(\mathbf{x}_i,\mathbf{m}_q)\), donc
Remplacement des anciens centres par les centres de gravité des groupes : si \(\widetilde{\mathbf{m}}_j\) est l’ancien centre du groupe \(j\) et \(\mathbf{m}_j\) le nouveau, alors
Comme \(\phi_\mathcal{E}(\mathcal{C}) \geq 0\), le processus itératif doit converger. La solution obtenue sera néanmoins un minimum local, dépendant de l’initialisation, souvent de valeur \(\phi_\mathcal{E}(\mathcal{C})\) bien plus élevée que celle correspondant au minimum global (inconnu).
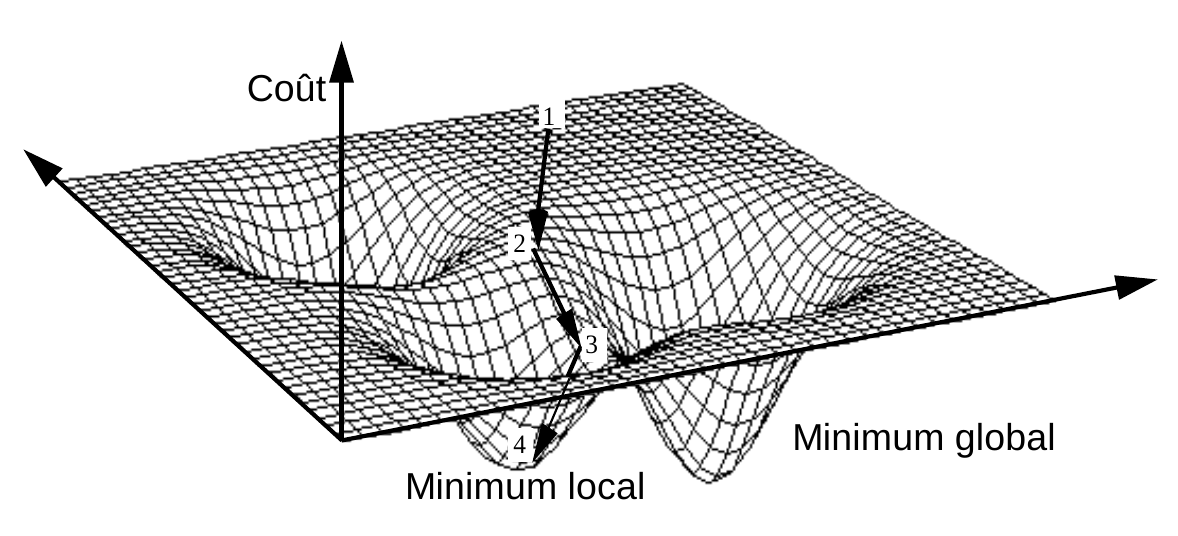
La figure suivante illustre le principe de la minimisation itérative d’une fonction différentiable de deux variables. Les variables sont ici associées aux axes horizontaux et la valeur de la fonction à minimiser à l’axe vertical. A partir de l’état initial 1, un minimum (local, dans cette figure) est atteint après 3 étapes.
Fig. 65 Minimisation itérative d’une fonction de deux variables, différentiable¶
Contrairement au cas illustré ci-dessus, la fonction \(\phi_\mathcal{E}(\mathcal{C})\)n’est pas différentiable (ni même continue) par rapport aux \(\mathbf{m}_j\) car un changement infinitésimal dans la position d’un centre peut provoquer un changement d’affectation de données aux centres et donc un changement significatif de la valeur de \(\phi_\mathcal{E}(\mathcal{C})\).
On appelle parfois k-moyennes (K-means) une version stochastique de la méthode des centres mobiles décrite ci-dessous (les entrées et les sorties sont les mêmes que pour l’algorithme précédent) :
Affectation de chaque donnée au groupe du centre le plus proche ;
Chaque groupe est initialisé avec son centre comme seul membre du groupe ;
tantque (centres non stabilisés) faire
Choix aléatoire d’une des données ;
(Ré)affectation de la donnée au groupe du centre le plus proche ;
Recalcul des centres pour le groupe rejoint par la donnée et le groupe quitté par la donnée ;
fintantque
Dans cette perspective, l’algorithme des centres mobiles est une variante « par lots » (batch) de K-means. Toutefois, K-means est souvent utilisé comme parfait synonyme de l’algorithme des centres mobiles ; c’est ainsi que ce terme sera employé dans la suite du cours. Vous trouverez ici un bref historique de K-means.
Noter qu’en général on emploie une variante intermédiaire dans laquelle à chaque itération on choisit un échantillon de \(b\) données (mini-batch) à (ré)affecter, avec \(b>1\).
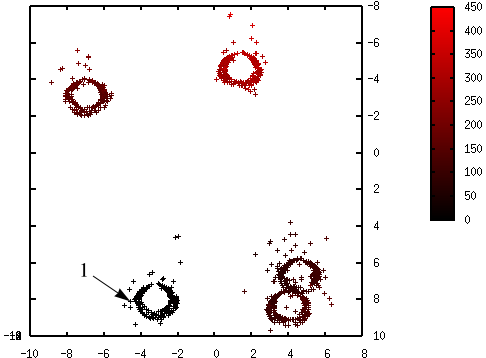
L’initialisation des centres est une étape critique de K-means. La figure suivante illustre les résultats obtenus avec K-means à partir de trois autres initialisations aléatoires des centres :
Fig. 66 Résultats différents avec initialisations aléatoires différentes de K-means¶
On constate que ces trois solutions sont sous-optimales : pour chacune, un des groupes « naturels » (on peut en juger visuellement car les données sont bidimensionnelles et peu nombreuses, ce ne sera pas le cas en général) est divisé en deux, et deux des groupes « naturels » sont regroupés. Les valeurs de \(\phi_\mathcal{E}(\mathcal{C})\) sont plus élevées pour ces trois résultats que pour le résultat optimal illustré plus haut. Exécuter K-means plusieurs fois, à partir d’initialisations aléatoires différentes, ne garantit pas de trouver la solution optimale (ou, dans le cas général, une solution proche de la solution optimale).
Une bonne initialisation de l’algorithme K-means doit permettre d’obtenir une solution de meilleure qualité mais aussi une convergence plus rapide (c’est à dire avec moins d’itérations) vers cette solution.
Parmi les nombreux algorithmes d’initialisation de K-means nous considérerons ici K-means++ proposé dans [AV07]. L’idée de départ est assez simple : choisir les centres l’un après l’autre, suivant une loi non uniforme qui évolue après le choix de chaque centre et qui privilégie les candidats éloignés des centres déjà sélectionnés. L’algorithme d”initialisationK-means++ est :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données de \(\mathbb{R}^p\) et nombre souhaité de centres \(k\) ;
Sorties : \(\mathcal{C} = \{\mathbf{c}_j, 1 \leq j \leq k\}\), à utiliser après comme centres de départ dans K-means ;
\(\mathcal{C} \leftarrow\) un \(\mathbf{x}\) de \(\mathcal{E}\) choisi au hasard ;
tantque (\(\|\mathcal{C}\| \le k\)) faire
Sélectionner \(\mathbf{x} \in \mathcal{E}\) avec la probabilité \(\frac{d^2(\mathbf{x},\mathcal{C})}{\phi_\mathcal{E}(\mathcal{C})}\) ;
Dans cet algorithme, nous avons noté \(d^2(\mathbf{x},\mathcal{C}) = \min_{j=1,\ldots,t} d^2(\mathbf{x},\mathbf{c}_j)\), la fonction (21) peut donc être écrite \(\phi_\mathcal{E}(\mathcal{C}) = \sum_{\mathbf{x} \in \mathcal{E}} d^2(\mathbf{x},\mathcal{C})\).
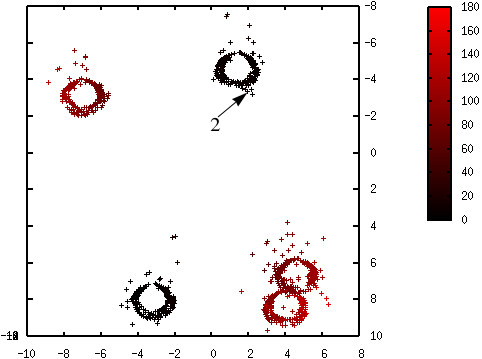
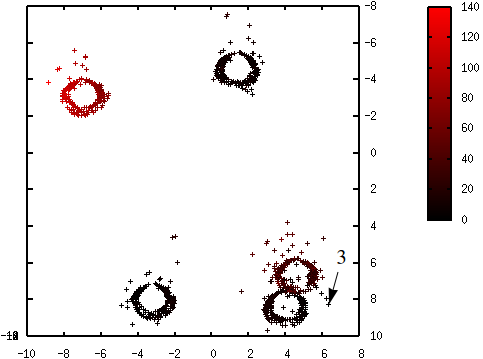
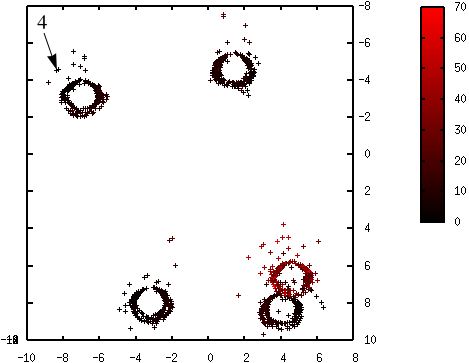
Les figures suivantes illustrent l’évolution des probabilités lors du déroulement de l’algorithme K-means++. La probabilité de sélection d’une donnée dans l’étape suivante (proportionnelle à \(d^2(\mathbf{x},\mathcal{C})\)) est d’autant plus élevée que la couleur est proche du rouge. La première coordonnée correspond à l’axe horizontal, la seconde à l’axe vertical.
Fig. 67 Probabilités après la sélection du premier centre (en bas à gauche) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{c}-4,6\\8,0\end{array}\right)\right\}\)¶
Fig. 68 Probabilités après la sélection du deuxième centre (en haut à droite) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{cc}-4,6 & 2,15\\8,0 & -3,45\end{array}\right)\right\}\)¶
Fig. 69 Probabilités après la sélection du troisième centre (en bas à droite) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{ccc}-4,6 & 2,15 & 6,32\\8,0 & -3,45 & 8,22\end{array}\right)\right\}\)¶
Fig. 70 Probabilités après la sélection du quatrième centre (en haut à gauche) : \(\mathcal{C} = \left\{\left(\begin{array}[h]{cccc}-4,6 & 2,15 & 6,32 & -8,37\\8,0 & -3,45 & 8,22 & -4,54\end{array}\right)\right\}\)¶
La sélection séquentielle des centres opérée par K-means++ permet de bien répartir les centres dans les données. Cela diminue le risque d’avoir, d’une part, des groupes de données sans aucun centre à proximité au démarrage de K-means et, d’autre part, plusieurs centres très proches entre eux. Ce sont ces insuffisances d’une initialisation aléatoire uniforme qui avaient provoqué la convergence vers des solutions sous-optimales dans les cas simples illustrés plus tôt Fig. 66.
Au-delà de sa simplicité conceptuelle, K-means présente d’autres intérêts qui justifient son utilisation fréquente. Il faut mentionner d’abord sa complexité algorithmique comparativement faible : \(O(t k N)\), \(t\) étant le nombre d’itérations, \(k\) le nombre de groupes (ou de centres) et \(N\) le nombre de données (observations). A première vue, la complexité serait donc linéaire en \(N\). Si on considère toutefois que le nombre \(k\) de groupes n’est pas fixé mais dépend de \(N\) (par ex. \(k \sim N^{1/3}\)) alors la complexité est légèrement sur-linéaire en \(N\) mais reste faible si on compare à d’autres méthodes. Il faut noter également que le nombre d’itérations nécessaire pour obtenir la convergence peut être fortement réduit avec une bonne initialisation, utilisant par ex. K-means++. Un autre intérêt est la présence d’un unique paramètre, qui est la valeur souhaitée pour le nombre de groupes (\(k\)). En revanche, les résultats peuvent dépendre fortement de la valeur de \(k\) (surtout pour un nombre faible de groupes) et la « bonne » valeur est difficile à choisir (voir plus loin).
K-means présente également un certain nombre de limitations, dont on peut parfois s’affranchir assez facilement.
Une limitation évidente est la nécessité de disposer de données vectorielles pour pouvoir calculer les centres des groupes. Cette limitation est levée dans la méthode des k-medoids qui se satisfait de l’existence d’une métrique (au prix d’une augmentation de la complexité algorithmique).
Une limitation plus profonde est liée au critère de regroupement. Comme pour K-means chaque groupe est représenté lors du déroulement de l’algorithme par un unique prototype (son centre), la méthode n’est pas adaptée à la découverte de groupes denses et non « compacts » séparés par des régions moins denses. Des extensions qui emploient plusieurs prototypes par groupe permettent de lever dans une certaine mesure cette limitation, mais le choix le plus fréquent dans une telle situation est d’avoir recours à une méthode très différente, comme la classification spectrale ou la classification sur la base de la densité (par ex. DBSCAN et algorithmes dérivés).
La limitation de K-means aux classes de forme globalement sphérique est due à l’utilisation de la distance euclidienne usuelle et peut être facilement levée par l’emploi d’autres métriques (comme celle de Mahalanobis), au prix de l’estimation de paramètres supplémentaires (pour la distance de Mahalanobis il faut calculer une matrice de covariances empiriques par groupe).
L’utilisation de la moyenne pour calculer les centres des groupes rend K-means sensible à la présence de données atypiques ou aberrantes. Cette sensibilité peut être réduite par une estimation robuste de la moyenne, l’emploi d’un critère à minimiser plus robuste ou des extensions floues de la méthode (comme fuzzy K-means). Les extensions floues présentent par ailleurs de meilleures propriétés de convergence et permettent d’obtenir des degrés d’appartenance de chaque donnée aux différents groupes.
Enfin, si le choix du nombre \(k\) de groupes a un fort impact sur les résultats et peut rarement être fait en amont de l’application de K-means, des méthodes existent pour sélectionner une bonne valeur pour \(k\) (voir plus loin).
L’emploi dans K-means des centres de gravité des groupes limite cette méthode aux données vectorielles. Dans de nombreux cas, nous souhaitons trouver des groupes dans des données pour lesquelles aucune représentation vectorielle convenable n’est disponible, par ex. des séquences de longueur variable, des sous-ensembles d’un très grand ensemble, des arbres ou des graphes. Lorsqu’une métrique (ou mesure de distance) est disponible pour ces données, il est facile d’adapter l’idée de K-means en remplaçant les centres de gravité des grupes par les « médoïdes » (medoids). Le médoïde d’un groupe est l’élément le plus « central » du groupe, c’est à dire celui pour lequel la somme des distances aux autres éléments du groupe est la plus faible.
Nous obtenons ainsi l’algorithme des K-medoids, dans lequel le seul changement par rapport à K-means est le remplacement des centres de gravité par des médoïdes :
Entrées : ensemble \(\mathcal{E}\) de \(N\) données, métrique \(d\) ;
Sorties : \(k\) groupes (clusters) disjoints \(\mathcal{E}_1,\ldots,\mathcal{E}_k\) et ensemble \(\mathcal{C}\) de leurs médoïdes ;
Initialisation aléatoire : choix aléatoire de \(k\) données comme médoïdes ;
tantque (medoids non stabilisés) faire
Choix, pour chaque donnée, du médoïde \(\mathbf{m}_{C(l)}\) le plus proche : \(C(l) = \arg \min_{j} d(\mathbf{x}_l, \mathbf{m}_j)\) ;
Constitution des groupes : \(\mathcal{E}_j\) est constitué de tous les \(\mathbf{x}_l\) qui sont plus proches de \(\mathbf{m}_j\) que de tout autre médoïde ;
Recherche des médoïdes de ces (nouveaux) groupes : \(\mathbf{m}_j = \arg \min_{\mathbf{x}_l \in \mathcal{E}_j} \sum_{\mathbf{x}_p \in \mathcal{E}_j} d(\mathbf{x}_l, \mathbf{x}_p)\) ;
fintantque
Cette version est issue de [HTF01]. Noter toutefois que l’algorithme cherche à minimiser un coût total qui est la somme des distances entre les données et les médoïdes des groupes auxquels ces données sont assignées. La première variante, Partitioning Around Medoids ([KR87]), est basée sur des essais successifs pour remplacer chaque médoïde par une autre donnée afin de réduire le coût total.
Comme pour K-means, l’initialisation a un impact important sur la qualité de la solution (du minimum local obtenu) et sur la rapidité de la convergence (le nombre d’itérations nécessaire). Il est conseillé d’employer une initialisation suivant le même principe que K-means++, c’est à dire la sélection séquentielle des centres (médoïdes ici) en maximisant l’éloignement de chaque nouveau centre des centres déjà choisis.
L’application de K-medoids à des données vectorielles, en utilisant la même métrique euclidienne que K-means, met en évidence une meilleure robustesse de K-medoids par rapport à K-means. Cette robustesse est le résultat de l’utilisation de médoïdes, moins affectés que les centres de gravité par les données excentrées.
En revanche, le remplacement des centres de gravité par des médoïdes présente un désavantage significatif en termes de complexité algorithmique. En effet, l’étape de recherche des médoïdes a pour conséquence une complexité \(O(N^2)\), alors que la complexité de K-means est plutôt \(O(N)\) (ou \(O(N^{4/3})\) si on tient compte du fait que le nombre de centres dépend de \(N\)).
Quelles que soient les données à regrouper, la plupart des algorithmes de classification automatique, dont K-means et K-medoids, convergent vers un résultat comportant en général des groupes. Quelle est la signification de ce résultat ? Y a-t-il réellement des groupes dans les données ? Leur nombre correspond ou non au nombre de groupes issu de l’algorithme ? Quel algorithme donne de meilleurs résultats sur mes données ? Nous abordons brièvement ces questions dans la suite.
Les méthodes de partitionnement que nous avons vues jusqu’ici ne modifient pas le nombre de groupes (\(k\)) fourni en entrée de l’algorithme. Comment choisir alors le « bon » nombre de groupes, vu que des informations permettant de le définir a priori sont très rarement disponibles ?
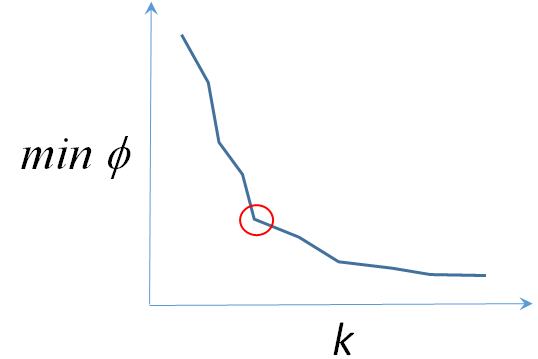
Nous avons également constaté que la valeur minimale que pouvait atteindre le critère (21) diminuait avec l’augmentation de \(k\) (et peut même arriver à \(0\) pour \(k = N\)), il n’est donc pas envisageable d’utiliser ce critère pour choisir entre plusieurs valeurs de \(k\). Parmi les différentes approches employées pour le choix de \(k\) nous pouvons mentionner :
La méthode du « coude ». Il est nécessaire d’appliquer l’algorithme de classification automatique (par ex. K-means avec initialisation par K-means++ ) pour un ensemble de valeurs du nombre de groupes \(k\) et de représenter sur un graphique les valeurs minimales atteintes par \(\phi_\mathcal{E}(\mathcal{C})\) (voir Fig. 71). La valeur retenue pour \(k\) est celle qui marque le début d’un pallier : pour des valeurs inférieures la qualité de regroupement est nettement moins bonne, alors que pour des valeurs supérieures la qualité ne s’améliore pas sensiblement. Noter toutefois que si la plage de valeurs de \(k\) explorées est suffisamment large il est fréquent d’y trouver plusieurs « coudes ».
Fig. 71 Choix du nombre \(k\) de groupes avec la méthode du coude¶
La reformulation du problème de classification automatique dans un cadre probabiliste. Cela permet de choisir un critère mieux fondé, par exemple un critère d’information comme AIC (Akaike) ou BIC (Bayes) pour identifier la valeur optimale pour \(k\), voir le chapitre sur l’estimation de densité.
L’étude de la stabilité des résultats. Des résultats relativement récents (voir par ex. [ST10]) proposent la stabilité comme critère de choix de modèle : par ex., une valeur de \(k\) est meilleure si les groupes obtenus sont plus stables à l’initialisation aléatoire de l’algorithme.
Les méthodes mentionnées permettent de choisir a posteriori une bonne valeur pour le nombre \(k\) de groupes au prix de calculs plus lourds, car l’algorithme de classification doit être appliqué une fois (ou même plusieurs fois afin d’évaluer la stabilité) pour chaque valeur candidate.
Les résultats obtenus par classification automatique sont d’autant plus significatifs que deux méthodes de classification différentes, ou deux initialisations différentes pour une même méthode sensible à l’initialisation, produisent (approximativement) les mêmes groupes. Pour mesurer la cohérence entre les résultats de deux applications différentes d’algorithmes de classification il est possible d’employer différentes mesures, par ex. (voir [WW07][VEB10]) :
L’indice de Rand ajusté. Une classification \(\mathcal{C}\) est définie comme l’ensemble des paires d’observations qui sont dans un même groupe, parmi toutes les paires possibles. On note par \(n_{11}\) le nombre de paires qui sont dans un même groupe suivant \(\mathcal{C}\) et \(\mathcal{C}'\), par \(n_{00}\) le nombre de paires qui sont dans des groupes différents suivant \(\mathcal{C}\) et \(\mathcal{C}'\), par \(n_{10}\) le nombre de paires dans un même groupe suivant \(\mathcal{C}\) et groupes différents suivant \(\mathcal{C}'\), et enfin par \(n_{01}\) le nombre de paires dans un même groupe suivant \(\mathcal{C}'\) et groupes différents suivant \(\mathcal{C}\).
Si \(\mathcal{C}, \mathcal{C}'\) sont deux classifications différentes, l’indice de Rand est la probabilité pour que les deux classifications soient en accord pour une paire de données choisie au hasard. Cela peut s’exprimer sous la forme suivante
où \(E(\mathcal{R})\) est l’espérance de l’indice de Rand et \(\max(\mathcal{R})\) la valeur maximale qu’il peut atteindre. Nous nous servirons de l’indice de Rand ajusté dans la séance de travaux pratiques.
où \(I(\mathcal{C},\mathcal{C}')\) est l’information mutuelle entre \(\mathcal{C},\mathcal{C}'\) et \(H(\mathcal{C})\), \(H(\mathcal{C}')\) les entropies respectives des deux partitionnements.
La qualité des résultats obtenus par une méthode de classification automatique peut être évaluée en examinant dans quelle mesure les groupes identifiés sont bien « ajustés » aux données. Par exemple, dans quelle mesure les données d’un même groupe sont plus proches entre elles que des données d’autres groupes.
De nombreux critères ont été proposés pour cela, comme le coefficient de silhouette (mesure à quel point les groupes sont « compacts » et bien séparés entre eux), la statistique modifiée de Hubert (mesure l’alignement entre distance et partitionnement), l’indice Davies-Bouldin (qui peut être vu comme un rapport entre inerties intra-groupes et inerties inter-groupes), etc. Plusieurs critères de ce type sont disponibles dans Scikit-learn. En général, ces critères ne peuvent pas être optimisés directement par un algorithme de classification automatique.
Il est important de noter que ces critères reflètent à la fois les propriétés des données elles-mêmes (forment-elles naturellement des groupes compacts et bien séparés) et l’ajustement aux données de la solution trouvée par l’algorithme de classification automatique. Cette caractéristique doit être prise en compte lorsqu’on compare les valeurs d’un même critère sur deux problèmes différents (c’est à dire avec des données différentes).
Nous pouvons remarquer que les critères (ou fonctions de coût) minimisés par les différentes méthodes de classification automatique (comme (21) pour K-means) reflètent également l’ajustement des groupes aux données mais ne permettent pas de comparer entre eux les résultats sur deux problèmes différents, ni de comparer à une référence.
Pour obtenir les meilleurs résultats de classification automatique sur un ensemble de données il est nécessaire d’appliquer plusieurs méthodes de classification différentes et de choisir ensuite celle qui mène aux meilleurs résultats. Les critères d’évaluation mentionnés dans la section précédente sont bien adaptés à ce type de comparaison car sur un même ensemble de données les différences mesurées concernent exclusivement l’ajustement des groupes trouvés aux données. En revanche, ces critères peuvent être plus ou moins adaptés aux données à traiter, par ex. le coefficient de silhouette n’est pas le critère adéquat pour des données qui se regroupent sur la base de la densité et non de la compacité.
Dans certains cas il est possible que des connaissances a priori concernant le regroupement ou non d’une partie des données soient disponibles. Par exemple, le fait que certaines données appartiennent à certains groupes (information non disponible pour tous les groupes et encore moins pour toutes les données), ou que certaines données doivent appartenir à un même groupe (contraintes must-link) ou à des groupes différents (contraintes cannot-link). Ces connaissances ne sont pas nécessairement directement exploitables dans un algorithme de classification (par ex. dans une fonctionnelle à minimiser), bien que des avancées récentes ont été faites dans le domaine de la classification semi-supervisée (semi-supervised clustering, voir par ex. [Bai13]). En revanche, ces connaissances peuvent toujours être employées à l’issue de la classification automatique pour évaluer les résultats obtenus.
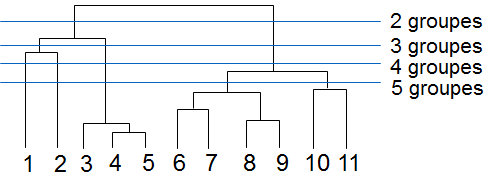
Parmi les méthodes qui visent à obtenir une hiérarchie de regroupements, nous examinons brièvement dans la suite la classification ascendante hiérarchique (CAH). Par rapport à un simple partitionnement des données, une hiérarchie de regroupements fournit une information plus riche concernant la structure de similarité des données. En effet, elle permet d’examiner l’ordre des agrégations de groupes, les rapports des similarités entre groupes, etc.
La classification ascendante procède par agrégations successives de groupes. A partir de la hiérarchie de groupes résultante il est possible d’observer l’ordre des agrégations de groupes, d’examiner les rapports des similarités entre groupes, ainsi que d’obtenir plusieurs partitionnements à des niveaux de granularité différents.
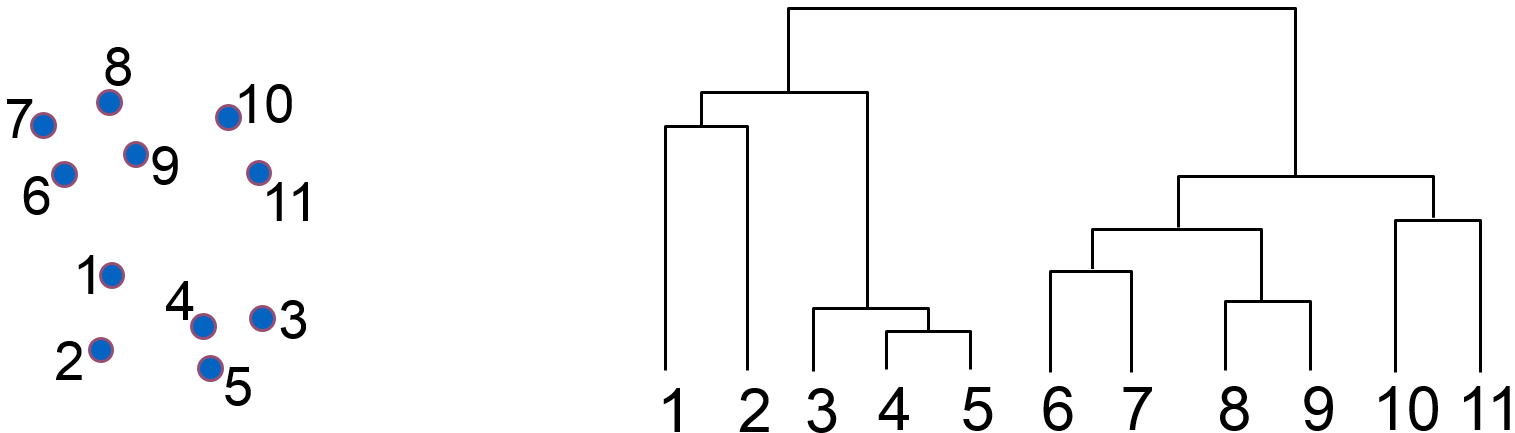
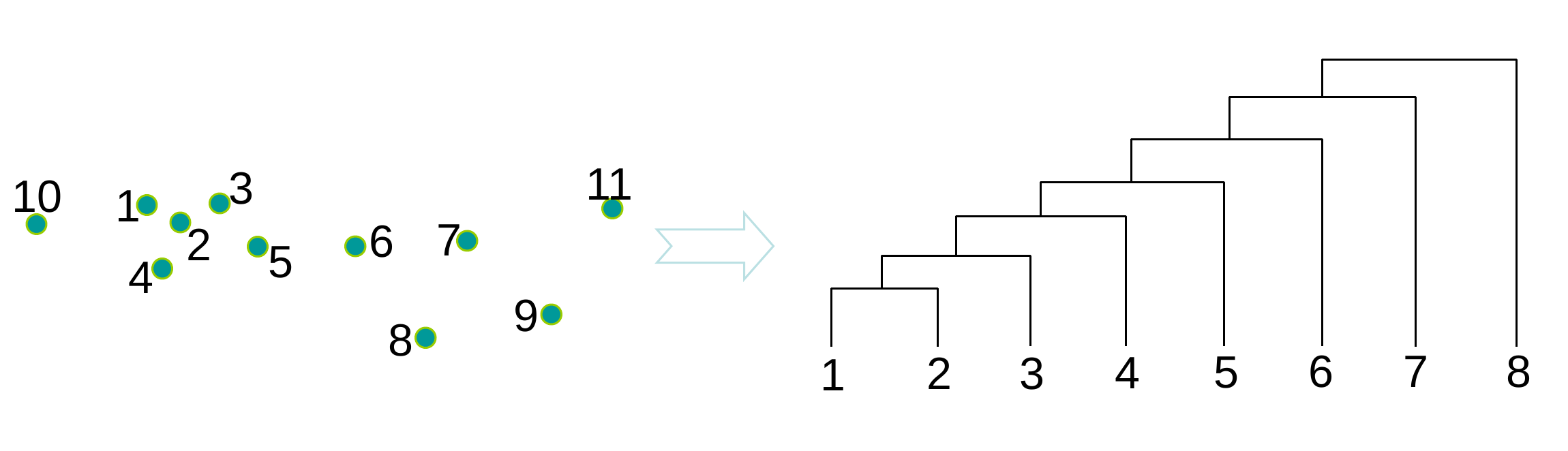
L’exemple suivant montre la hiérarchie de groupes (« dendrogramme ») obtenue par agrégations successives à partir d’un petit ensemble de données bidimensionnelles :
Fig. 72 Dendrogramme (à droite) obtenu à partir des données 2D (à gauche)¶
Le principe de la classification ascendante est de regrouper (ou d’agréger), à chaque itération, les données et/ou les groupes les plus proches qui n’ont pas encore été regroupé(e)s.
Considérons un ensemble de données \(\mathcal{E} \subset \mathcal{X}\) et une distance \(d_\mathcal{X} : \mathcal{X} \times \mathcal{X} \rightarrow \mathbb{R}^+\). Les données individuelles peuvent être comparées grâce à cette distance. Mais, une fois deux données agrégées dans un groupe (noté \(h_p\), par ex.), comment mesurer la similarité de ce groupe à une autre donnée ou à un autre groupe (\(h_q\), par ex.) ? Il est nécessaire de définir une nouvelle mesure pour la dissimilarité entre groupes (ou entre une donnée et un groupe). Cette mesure est en général appelée « indice d’agrégation » et plusieurs choix fréquents sont :
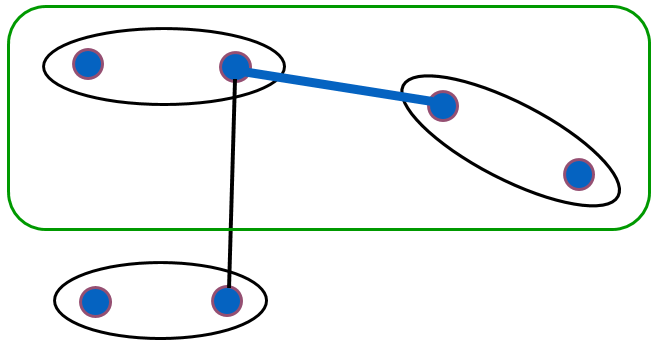
Le lien minimum (single linkage) : l’indice d’agrégation entre deux groupes \(h_p,h_q\) est la valeur la plus faible des distances entre une donnée du premier groupe et une donnée du second. Formellement, \(\delta_s(h_p,h_q) = \min_{x_i \in h_p, x_j \in h_q} d_\mathcal{X}(x_i,x_j)\). Il suffit donc que deux groupes contiennent deux éléments proches pour que la valeur de l’indice soit faible :
Fig. 73 Illustration du lien minimum (single linkage)¶
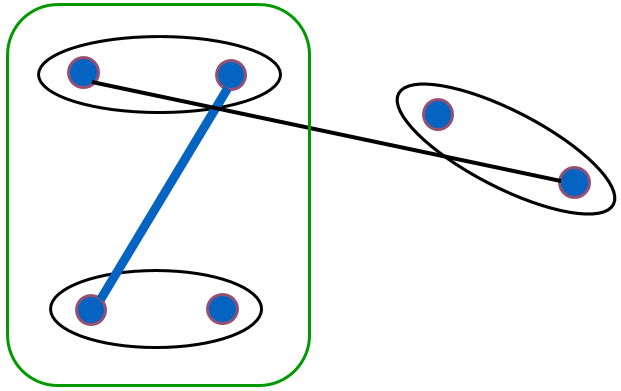
Le lien maximum (complete linkage) : l’indice d’agrégation entre deux groupes \(h_p,h_q\) est la valeur la plus élevée des distances entre une donnée du premier groupe et une donnée du second (appelée parfois « diamètre » de l’agrégat). Formellement, \(\delta_s(h_p,h_q) = \max_{x_i \in h_p, x_j \in h_q} d_\mathcal{X}(x_i,x_j)\). Il est donc nécessaire que tous les éléments d’un des groupes soient proches de tous les éléments de l’autre groupe pour que la valeur de l’indice soit faible :
Fig. 74 Illustration de l’agrégation utilisant le lien maximum (complete linkage)¶
Le lien moyen (average linkage) : l’indice d’agrégation entre deux groupes \(h_p,h_q\) est la moyenne des distances entre une donnée du premier groupe et une donnée du second. Formellement, \(\delta_s(h_p,h_q) = \frac{1}{\|h_p\|\cdot\|h_q\|} \sum_{x_i \in h_p, x_j \in h_q} d_\mathcal{X}(x_i,x_j)\) :
Fig. 75 Illustration de l’agrégation utilisant le lien moyen (complete linkage)¶
Les illustrations précédentes représentent des données de \(\mathbb{R}^2\), mais les définitions des trois indices montrent bien que la seule exigence est l’existence d’une distance entre données individuelles et non d’une structure d’espace vectoriel.
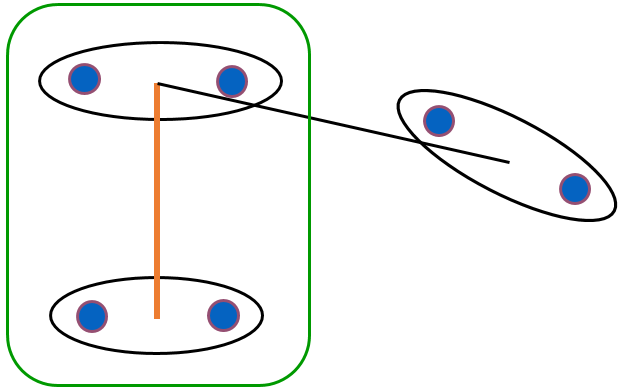
L’indice de Ward, en revanche, est défini uniquement pour des données vectorielles. La valeur de l’indice correspond à l’augmentation de l’inertie intra-groupe lors de l’agrégation de deux groupes. Elle est la différence entre l’inertie intra-groupe de l’agrégat et la somme des inerties intra-groupe des deux groupes agrégés. Formellement, \(\delta_s(h_p,h_q) = \frac{\|h_p\|\cdot\|h_q\|}{\|h_p\|+\|h_q\|} d^2_\mathcal{X}(\mathbf{m}_p,\mathbf{m}_q)\).
Fig. 76 Illustration de l’agrégation utilisant l’indice de Ward¶
Contrairement à l’algorithme K-means, qui exige des données vectorielles, la CAH se satisfait, pour de nombreux indices d’agrégation (par ex. lien minimum, lien maximum, lien moyen) d’une simple distance définie sur l’espace des données. Il est donc possible de l’employer directement pour des données qui n’ont pas de représentation vectorielle naturelle.
L’algorithme de CAH est très simple : on agrège, lors d’itérations successives, les groupes entre lesquels l’indice d’agrégation choisi a la valeur minimale.
Entrées : \(\mathcal{E}\) de \(N\) données de \(\mathcal{X}\) muni de la distance \(d_\mathcal{X}\), indice d’agrégation \(\delta_s\) ;
Sorties : Hiérarchie de groupes (dendrogramme) ;
Chaque donnée définit un groupe ;
tantque (nombre de groupes \(> 1\)) faire
Calcul des valeurs de l’indice d’agrégation entre tous les groupes issus de l’itération précédente ;
Regroupement des 2 groupes ayant la plus petite valeur de l’indice d’agrégation ;
fintantque
Dans la hiérarchie de regroupements (dendrogramme) illustrée plus haut Fig. 72, la hauteur de chaque palier (qui représente une agrégation) est égale à la valeur de l’indice d’agrégation entre les groupes agrégés.
La complexité algorithmique pour un choix non contraint de l’indice d’agrégation est \(O(N^2 \log N)\). En revanche, pour l’indice du lien minimum et l’indice du lien maximum existent des algorithmes de complexité \(O(N^2)\), complexité qui reste néanmoins élevée. Une pratique assez fréquente est d’appliquer d’abord l’algorithme K-means avec une valeur élevée de \(k\) (mais néanmoins \(k \ll N\)), ensuite d’utiliser la classification ascendante hiérarchique pour regrouper les \(k\) groupes issus de K-means.
Le choix de l’indice d’agrégation a un impact parfois significatif sur les résultats obtenus. Au-delà des contraintes liées à la nature des données (éléments d’un espace vectoriel ou simplement métrique), une bonne compréhension du problème doit aider ce choix. Par exemple, l’utilisation du lien minimum mène à un résultat qui se rapproche du regroupement par densité, alors que l’emploi du lien maximum (ou de l’indice de Ward) produit plutôt des groupes « compacts » séparés entre eux. En revanche, le lien minimum mène souvent à des arbres en escalier, déséquilibrés et peu exploitables, comme dans l’exemple suivant :
Fig. 77 Illustration de l’effet escalier obtenu avec l’indice du lien minimum¶
Quelle que soit la hauteur à laquelle on coupe un tel arbre, le partitionnement obtenu n’est pas exploitable car le résultat correspond à un grand groupe plus un ensemble de données individuelles.
Comme nous l’avons vu précédemment, un inconvénient important de l’algorithme des k-moyennes est son hypothèse fondamentale : on suppose que les groupes recherchés sont compacts et donc resserrés autour de leur centre.
Or il est fréquent que les groupes « naturels » soient de forme arbitraire. Le critère de regroupement n’est donc plus la distance au centre du groupe mais la proximité à d’autres points du groupe. Autrement dit, le critère qui permet de définir un groupe est la densité des points qui le constituent.
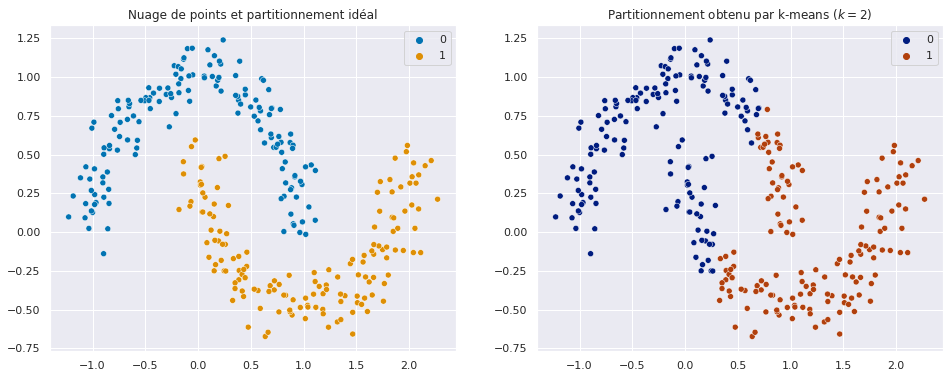
Fig. 78 Illustration du partitionnement obtenu par k-means lorsque l’hypothèse de compacité n’est pas respectée.¶
L’illustration ci-dessus Fig. 78 est particulièrement parlante : k-means regroupe des points issus des deux demi-lunes différentes. Ces points ont beau être proches, l’existence d’une zone « vide » entre eux laisse suggérer qu’ils n’appartiennent pas réellement au même ensemble.
Ainsi, les algorithmes de classification automatique basés sur la densité font l’hypothèse suivante : les groupes sont constitués d’ensemble de points denses séparés par des régions de faible densité. Intuitivement il faut imaginer que les groupes forment un archipel constitué d’îlots de points resserrés au milieu d’un océan distendu.
Mathématiquement, on définira la densité comme le nombre de points dans une boule d’un certain rayon \(r\) (qui sera un paramètre de l’algorithme).
Considérons un espace \(\mathcal{X}\) doté d’une métrique \(d\) (typiquement, \(\mathcal{X}\) est un espace vectoriel mais ce n’est pas indispensable). Soit \(\mathcal{D} = \{\mathbf{x}_1, \dots, \mathbf{x}_N\} \subset \mathcal{X}\) un jeu de données (d’observations). On définit l”\(\varepsilon\)-voisinage \(V_\varepsilon(\mathbf{x})\) du point \(\mathbf{x} \in \mathcal{X}\) comme étant l’ensemble des points de \(\mathcal{D}\) se trouvant à une distance inférieure à \(\varepsilon\) de \(\mathbf{x}\) :
Autrement dit, l”\(\varepsilon\)-voisinage d’un point \(\mathbf{x}\) est l’intersection de la boule (ouverte) de \(X\) de rayon \(\varepsilon\) centrée sur \(\mathbf{x}\) et du jeu de données \(\mathcal{D}\).
On dira qu’un \(\varepsilon\)-voisinage est \(m\)-dense si la cardinalité du voisinage est supérieure ou égale à \(m \in \mathbb{N}^+\), c’est-à-dire qu’il y a au moins \(m\) points de \(\mathcal{D}\) dans la boule de rayon \(\varepsilon\). On considère ici que le point \(\mathbf{x}\) fait partie de son \(\varepsilon\)-voisinage et est donc compté dans la cardinalité.
Un algorithme de base en classification automatique par densité est l’algorithme DBSCAN (density-based spatial clustering of applications with noise) [EKS96]. Comme son nom l’indique, DBSCAN utilise la notion de densité pour partitionner les données, tout en étant robuste au bruit.
A chaque observation \(\mathbf{x}\) du jeu de données, DBSCAN assigne un des trois types suivants :
point central (core point) si son voisinage est \(m\)-dense, c’est-à-dire si le nombre de points dans la boule de rayon \(\varepsilon\) centrée sur \(\mathbf{x}\) contient au moins \(m\) points,
point frontière (border point) si ce n’est pas un point central mais qu’il appartient au voisinage d’un tel point, c’est-à-dire qu’il existe un point central dans la boule de rayon \(\varepsilon\) centrée sur \(\mathbf{x}\),
point « aberrant » ou bruit (noise point) ou point isolé si ce n’est ni un point central, ni un point frontière.
Lorsque l’on trouve un point central, on lui assigne une étiquette de groupe. Tous les points dans son voisinage (qui sont nécessairement soit des points centraux, soit des points frontière) reçoivent la même étiquette. On répète le processus pour tous les points centraux du voisinage.
Observons que l’algorithme DBSCAN ne nécessite à aucun moment de préciser à l’avance le nombre de groupes recherchés ! Cette propriété est un des avantages de DBSCAN, un autre avantage étant la robustesse au bruit. En effet, les points isolés ne sont assignés à aucun groupe (contrairement à k-means) et peuvent donc être « rejetés » de l’analyse.
Une version synthétique abstraite de DBSCAN serait (voir [SSE17]) :
En pseudo-code plus détaillé, l’algorithme de DBSCAN peut être écrit :
DBSCAN(D,epsilon,m)pourchaquepointPnonvisitédujeudedonnéesDmarquerPcommevisitéVoisins=voisinage(D,P,epsilon)si|Voisins|<=mmarquer(potentiellementprovisoirement)Pcommebruit(ouisolé)sinoncréerunnouveaugroupeGvidepropagerCluster(D,P,epsilon,Voisins,G,m)propagerCluster(D,P,epsilon,Voisins,G,m)ajouterPaugroupeGpourchaquepointQdeVoisinssiQn'a pas été visitémarquerQcommevisitéVoisins' = voisinage(D, Q, epsilon)si|Voisins'| >= mVoisins=VoisinsUVoisins'siQn'est pas dans un groupeajouterQaugroupeGvoisinage(D,P,epsilon)retournerlespointsdeDquisontàunedistanceinférieureàepsilondeP
Noter qu’un point marqué au départ comme bruit (ou point isolé) peut être trouvé ultérieurement (dans propagerCluster) comme point frontière car dans le voisinage d’un point central et ainsi être ajouté à un groupe.
La complexité algorithmique peut s’exprimer par le nombre d’opérations élémentaires, en l’occurrence de calculs de la distance entre deux points. Elle est dominée par les appels à la fonction voisinage. L’implémentation naïve, qui consiste à tester tous les autres points, est de complexité linéaire \(O(N)\). On utilise en général un index multidimensionnel (comme un ball tree, une des solutions employée dans Scikit-learn) afin de réaliser cette opération en complexité logarithmique \(O(\log N)\) en moyenne. L’obtention du voisinage s’effectue une fois pour chaque point, ce qui donne une complexité globale en moyenne de \(O(N \log N)\) et quadratique \(O(N^2)\) dans le pire des cas. Noter que les indexes multidimensionnels perdent en efficacité lorsque la dimension augmente (nous pouvons les considérer inefficaces pour des vecteurs de dimension supérieure à 10).
\(\varepsilon\) qui définit la taille du voisinage (le rayon de la boule),
\(m\) qui définit la densité minimale à atteindre (la cardinalité minimale de la boule pour que le point soit considéré central).
Ces paramètres peuvent être réglés à l’aide de connaissances expertes sur les données mais cela n’est pas toujours possible si l’on ne maîtrise pas bien les données.
Le choix de \(\varepsilon\) est le plus difficile. Si \(\varepsilon\) est choisi trop faible, alors peu de points seront considérés comme voisins (centraux ou frontière). La plupart des points seront considérés comme isolés, ce qui n’est pas informatif. À l’inverse, choisir une valeur trop élevée de \(\varepsilon\) rendra presque tous les points voisins les uns des autres, ce qui réduira le nombre de groupes obtenus. Dans le cas extrême, DBSCAN regroupe tous les points dans un même groupe, ce qui n’est pas non plus utile.
En pratique, on souhaite choisir la valeur la plus faible possible pour \(\varepsilon\). Cela permet d’avoir la meilleure tolérance au bruit. Une règle usuelle est d’inspecter le plus grand cluster obtenu avec DBSCAN : si celui-ci regroupe une trop grande partie du jeu de données (par exemple, au-delà de 30%), alors il faut réduire \(\varepsilon\) pour le sous-diviser.
Il existe toutefois deux heuristiques pour choisir \(\varepsilon\) ou \(m\), du moment que l’autre des deux paramètres est fixé (plus de détails sur les bonnes pratiques dans l’usage de DBSCAN se trouvent dans [SSE17]).
Remarquons que, si le nombre minimal de points dans le voisinage est de 1, alors tous les points sont centraux quel que soit le rayon \(\varepsilon\) du voisinage (car chaque boule de rayon \(\varepsilon > 0\) centrée sur un point contient au moins le point central). Dans le cas \(m = 2\), DBSCAN dégénère en un cas particulier de la classification ascendante hiérarchique utilisant le lien minimum, dont on tronque le dendrogramme à hauteur \(\varepsilon\). Par défaut, la plupart des implémentations de DBSCAN utilisent \(m = 4\) (proposé au départ pour des données bidimensionnelles) tandis que [SEK98] recommande simplement de fixer \(m\) comme étant le double de la dimensionnalité des données : \(m = 2 \cdot d\) où \(\mathbf{x}_i \in \mathbb{R}^d\).
Une autre heuristique consiste à déterminer, une fois la taille \(\varepsilon\) du voisinage choisie, la valeur \(m\) du nombre minimal de points permettant de définir un voisinage dense en calculant la densité moyenne :
Le rayon \(\varepsilon\) du voisinage caractéristique dans DBSCAN est le paramètre central de l’algorithme. Sa valeur dépend toutefois du jeu de données.
Une heuristique classique consiste à observer le graphe des \(k\)-distances du jeu de données. La \(k\)-distance d’un point \(\mathbf{x} \in \mathcal{D}\) du jeu de données correspond à la distance entre \(\mathbf{x}\) et son \(k\)-ième voisin le plus proche. Cela correspond ainsi au rayon de la plus petite boule qui englobe \(\mathbf{x}\) et ses \(k\) plus proches voisins.
Le graphe des \(k\)-distances du jeu de données \(\mathcal{D}\) représente les \(k\)-distances pour tous les points du jeu de données, ordonnées par valeur décroissante.
Pour un nombre de points dans le voisinage \(m\) fixé, on trace ainsi le graphe des \(k\)-distances triées en ordre décroissant, pour \(k = m - 1\). La courbe obtenue est de la forme ci-dessous:
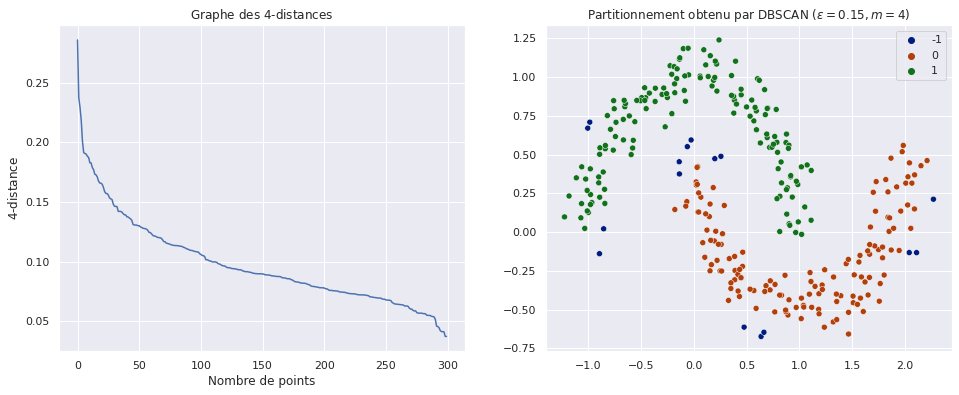
Fig. 79 Détermination du rayon de DBSCAN par la méthode du coude. A gauche, le graphe des \(k\)-distances calculées pour tous les points et triées en ordre décroissant. A droite, le partitionnement obtenu correspond à nos attentes : les deux demi-lunes sont bien séparées et les points isolés (de couleur bleu foncé, étiquette “-1”) sont rejetés.¶
La portion qui précède le « coude » correspond à des points dont les \(k\) plus proches voisins sont plutôt éloignés : il s’agit des points isolés. Dans la seconde portion du graphe la décroissance des \(k\)-distances est plus lente, nous trouvons là les points frontière et centraux, qui sont relativement bien connectés les uns aux autres et donc à des distances similaires. On choisit par exemple dans ce cas \(\varepsilon \approx 0.1 \sim 0.2\), ce qui permet d’obtenir le partionnement à droite de la figure.
Pour terminer, notons que, comme pour les k-moyennes, le choix de la distance \(d\) est crucial dans l’application de DBSCAN. Le choix de la métrique définit la notion de voisinage et il est important de choisir une distance adaptée à ses données. La distance euclidienne est la plus communément employée mais ces algorithmes s’appliquent avec n’importe quelle distance. Le choix de la métrique utilisée précède l’estimation des paramètres \(\varepsilon\) et \(m\), car la valeur de \(\varepsilon\) en dépend fortement.
Ces éléments mathématiques peuvent être ignorés en première lecture.
On dit qu’un point \(\mathbf{x}'\) est directement accessible par \(\varepsilon\)-densité depuis un point \(\mathbf{x}\) si le voisinage de \(\mathbf{x}\) est \(m\)-dense et que \(\mathbf{x}'\) fait partie de celui-ci, c’est-à-dire :
\(\mathbf{x}' \in V_\varepsilon(\mathbf{x})\),
\(V_\varepsilon(\mathbf{x})\) est \(m\)-dense.
On définit alors la notion d’accessibilité par \(\varepsilon\)-densité : \(\mathbf{x}'\) est accessible par densité depuis \(\mathbf{x}\) si et seulement si il existe une séquence \((\mathbf{y}_1, \dots, \mathbf{y}_n)\) telle que :
\(\mathbf{y}_1 = \mathbf{x}\) et \(\mathbf{y}_n = \mathbf{x}'\)
\(\mathbf{y}_{i+1}\) est directement accessible par densité depuis \(\mathbf{y}_i\)
Autrement dit, il existe une chaîne de points permettant d’aller de \(\mathbf{x}\) à \(\mathbf{x}'\) en passant exclusivement par des voisinages \(m\)-denses.
Enfin, on considère que \(\mathbf{x}\) et \(\mathbf{x}'\) sont densément connectés s’il existe \(\mathbf{o} \in D\) tel que \(\mathbf{x}\) et \(\mathbf{x}'\) sont accessibles par densité depuis \(\mathbf{o}\).
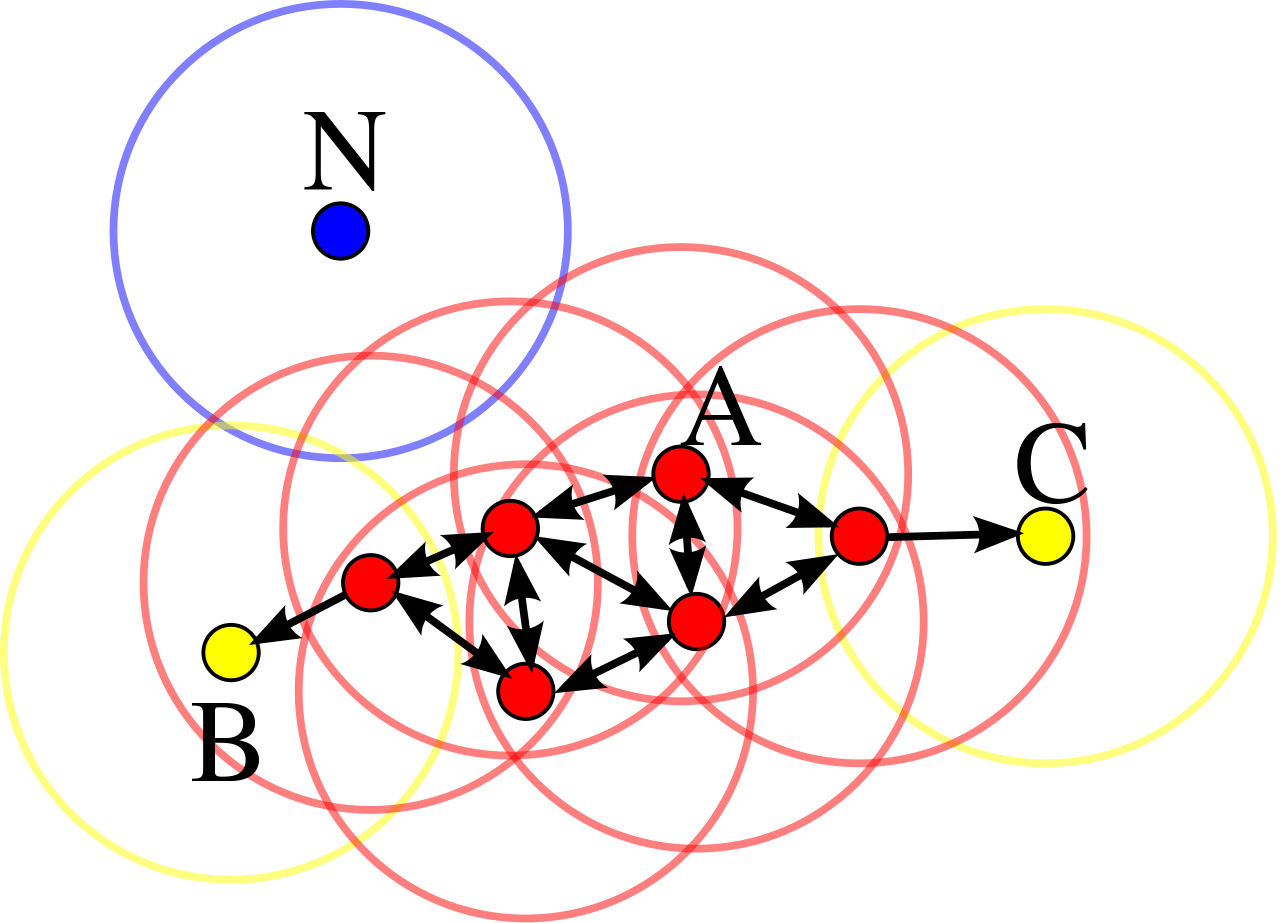
En réalité, DBSCAN ne fait que relier entre eux les points densément connectés. Plus spécifiquement, le partitionnement réalisé par DBSCAN correspond à créer un graphe dont les nœuds sont les points du jeu de données et les arêtes (orientées) représentent l’accessibilité par densité. Deux points font partie du même groupe si et seulement si ils sont densément connectés, c’est-à-dire s’ils font partie de la même composante connexe. Un point aberrant est un nœud isolé du graphe ainsi formé. Un exemple est donné dans la figure suivante :
Fig. 80 Graphe d’accessibilité par densité. Les points rouges sont des points centraux (ici \(m = 3\)), les points jaunes sont des points frontière et le point bleu est un point isolé.¶
Arthur, D. and S. Vassilvitskii. K-means++: The advantages of careful seeding. In Proceedings of the 18th Annual ACM-SIAM Symposium on Discrete Algorithms, SODA’07, pages 1027–1035, Philadelphia, PA, USA, 2007. Society for Industrial and Applied Mathematics.
Bouveyron, C., C. Brunet-Saumard. Model-based clustering of high-dimensional data: A review, Computational Statistics and Data Analysis, Volume 71, March 2014, Pages 52-78, ISSN 0167-9473, http://dx.doi.org/10.1016/j.csda.2012.12.008.
Ester, M., H.-P. Kriegel, J. Sander, and X. Xu. A density-based algorithm for discovering clusters in large spatial databases with noise, Proceedings of the 2nd International Conference on Knowledge Discovery and Data mining, 226232, 1996.
Jain, A. K., M. N. Murty, and P. J. Flynn. Data clustering: a review. ACM Comput. Surv. 31, 3 (September 1999), 264-323. DOI=http://dx.doi.org/10.1145/331499.331504.
Kaufman, L. and Rousseeuw, P.J. Clustering by means of Medoids, Statistical Data Analysis Based on the :math:`L_1` Norm and Related Methods, ed. Y. Dodge, North-Holland, 1987, pp. 405–416.
Sander, J., Ester, M., Kriegel, H.-P., Xu, X. Density-Based Clustering in Spatial Databases: The Algorithm GDBSCAN and Its Applications. Data Mining and Knowledge Discovery, Springer (1998), 169-194.
Schubert, E., Sander, J., Ester, M., Kriegel, H.-P., Xu, X., DBSCAN Revisited, Revisited: Why and How You Should (Still) Use DBSCAN. ACM Trans. Database Syst, 2017.
Vinh, N.X., J. Epps, and J. Bailey. Information theoretic measures for clusterings comparison: Variants, properties, normalization and correction for chance. J. Mach. Learn. Res., 11:2837–2854, Dec. 2010.