L’estimation directe du risque espéré par le risque empirique (appelée aussi estimation in-sample) est donc excessivement optimiste, surtout pour des familles \(\mathcal{F}\) de capacité élevée (ou infinie), et doit donc être évitée. Lorsque la capacité de \(\mathcal{F}\) est faible et le nombre \(N\) de données d’apprentissage très élevé (situation très rare en pratique), des bornes de généralisation \(B(N,\mathcal{F})\) suffisamment réduites pour majorer de façon utile le risque espéré peuvent éventuellement être obtenues et nous pouvons nous servir de \(R(f^*_{\mathcal{D}_N}) \leq R_{\mathcal{D}_N}(f^*_{\mathcal{D}_N}) + B(N,\mathcal{F})\).
Une méthode générale pour estimer le risque espéré est celle des données de test ou de l”échantillon-test (appelée aussi estimation out-of-sample) :
L’ensemble de données disponibles \(\mathcal{D}_N\) est partitionné en deux ensembles mutuellement exclusifs par sélection aléatoire, les données d’apprentissage \(\mathcal{A}\) (par ex. env. 70% du nombre total) et les données de validation \(\mathcal{V}\) (par ex. 30% du nombre total).
L’apprentissage du modèle est réalisé sur les données de l’ensemble \(\mathcal{A}\), en utilisant une des approches mentionnées (la MRE ou la MRER).
Le risque espéré du modèle résultant est estimé sur les données de \(\mathcal{V}\).
Apprendre sur une partie seulement des données disponibles (\(\mathcal{A}\)) peut être un inconvénient sérieux lorsque \(N\) est faible. Certaines classes peuvent être relativement rares dans les données, diminuer encore le nombre d’observations appartenant à ces classes a un impact négatif sur la qualité du modèle résultant.
Aussi, l’estimateur ainsi obtenu pour le risque espéré a une variance élevée : un autre découpage de \(\mathcal{D}_N\) en \(\mathcal{A}'\) et \(\mathcal{V}'\) peut produire un résultat assez différent. Afin de réduire la variance de l’estimateur il faudrait moyenner les résultats issus de plusieurs découpages différents de \(\mathcal{D}_N\), c’est ce que proposent les méthodes de validation croisée.
Avant d’examiner de plus près ces méthodes, il est important de rappeler qu’un problème de validité important rencontré dans la pratique est la non stationnarité des phénomènes modélisés : les données d’apprentissage deviennent de moins en moins représentatives au fil du temps car la loi conjointe inconnue \(P\) évolue. Il est donc important d’estimer régulièrement l’erreur sur des données récentes afin de mettre en évidence une éventuelle divergence du modèle et donc la nécessité de le mettre à jour.
Pour réduire la variance de l’estimation du risque espéré obtenue sur un échantillon-test, plusieurs partitionnements différents de \(\mathcal{D}_N\) en \(\mathcal{A}_i\) et \(\mathcal{V}_i\) sont réalisés, avec \(i \in {1,\ldots,k}\), \(\mathcal{D}_N = \mathcal{A}_i \cup \mathcal{V}_i\) et \(\mathcal{A}_i \cap \mathcal{V}_i = \varnothing\), à chaque fois un modèle \(f_i\) est appris sur \(\mathcal{A}_i\), son erreur \(L(\mathcal{V}_i,f_i)\) est calculée sur \(\mathcal{V}_i\), et enfin le risque espéré est estimé par la moyenne \(\frac{1}{k} \sum_{i=1}^k L(\mathcal{V}_i,f_i)\). Nous obtenins ainsi un estimateur de variance plus faible, tout en utlisant mieux les données disponibles.
Suivant les partitionnements réalisés, les méthodes peuvent être
Exhaustives, lorsque tous les partitionnements possibles respectant certains effectifs sont utilisés :
Leave\(p\)out (LPO) : \(N-p\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et \(p\) pour la validation (\(\mathcal{V}_i\)), tous les \(C_N^p\) partitionnements possibles avec ces effectifs sont utilisés. Il est donc nécessaire d’apprendre \(C_N^p\) modèles différents, ce qui implique en général un coût excessif.
Leave one out (LOO) : \(N-1\) données sont employées pour l’apprentissage (\(\mathcal{A}_i\)) et une seule pour la validation (\(\|\mathcal{V}_i\| = 1\)), il y a donc \(C_N^1 = N\) partitionnements possibles et il faut apprendre \(N\) modèles différents. Le coût reste élevé pour des données volumineuses (\(N\) élevé).
Non exhaustives :
k-fold : l’ensemble \(\mathcal{D}_N\) est partitionné en \(k\) parties, chaque modèle est évalué sur une des partitions et les \(k-1\) autres sont employées pour son apprentissage. Sont appris \(k\) modèles, chacun évalué sur une partition différente parmi les \(k\). Souvent \(k = 5\) ou \(k = 10\), mais le choix de \(k\) dépend des ressources disponibles car le coût augmente linéairement avec \(k\). La méthode leave one out peut être vue aussi comme un cas particulier de k-fold, pour \(k = N\).
Échantillonnage répété (shuffle and split) : un échantillon aléatoire de \(n\) données est utilisé pour la validation, les autres \(N-n\) données étant employées pour l’apprentissage du modèle, on répète cela \(k\) fois pour obtenir \(k\) modèles. Cette méthode permet d’éviter la contrainte \(k \cdot n = N\), mais présente le désavantage de trouver certaines données dans plusieurs ensembles de validation différents alors que d’autres ne seront présentes dans aucun échantillon.
Même les méthodes non exhaustives sont coûteuses, car il est nécessaire d’apprendre (et ensuite évaluer) \(k\) modèles plutôt qu’un seul. Il faut toutefois noter que ces \(k\) apprentissages (et les \(k\) évaluations correspondantes) peuvent être réalisées en parallèle si nous disposons d’une plate-forme adaptée.
L’estimateur ainsi obtenu pour le risque espéré est asymptotiquement (lorsque \(k = N\) tend vers l’infini) sans biais. Pour \(k = N\) fini, cet estimateur présente néanmoins un léger biais : il surestime le risque espéré car l’apprentissage se fait à chaque fois sur moins de \(N\) données. Le biais sera ainsi plus faible pour leave one out que pour k-fold. La variance de l’estimateur reste relativement élevée, en partie à cause de l’utilisation de relativement peu de données pour évaluer chaque modèle. On considère que la validation croisée présente un bon compromis entre le biais et la variance de l’estimateur.
On notera donc que la procédure k-fold est moins coûteuse que celle de leave one out car seulement \(k \ll N\) modèles sont appris et non \(N\) (pour \(k \ll N\)).
Ceci au prix d’un biais supérieur car pour k-fold l’apprentissage se fait à chaque fois sur \(\frac{N (k-1)}{k}\) données \(\left(\frac{N (k-1)}{k} < N-1\right)\),
Une lecture attentive aura permis de remarquer que, pour répondre à l’objectif d’estimation du risque espéré pour un modèle, nous avons choisi d’en développer (et évaluer) \(k\). Pour lequel de ces modèles est valide cette estimation du risque espéré ? En général, afin de traiter un problème de modélisation, avec un même ensemble \(\mathcal{D}_N\) sont employées plusieurs « procédures » de modélisation, chacune définie par une famille \(\mathcal{F}\), un critère de régularisation \(G(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc. La validation croisée sert ensuite à choisir, entre ces « procédures » de modélisation, celle pour laquelle le risque espéré estimé a la valeur la plus faible. La variance de l’estimateur restant relativement élevée, ce choix n’est pas nécessairement optimal. La « procédure » ainsi choisie est enfin employée pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). C’est à ce modèle que s’applique l’estimation du risque espéré obtenue pour la « procédure » dont le modèle est le résultat. Nous reviendrons plus loin sur les questions de sélection de modèle (ou de « procédure » de modélisation).
L’utilisation de la validation croisée impose, dans certains cas, des précautions :
Pour les problèmes de classement avec des classes déséquilibrées (certaines classes sont bien plus rares que d’autres) : afin d’être assuré de conserver les rapports entre les classes dans tous les découpages, il est nécessaire d’utiliser
Un partitionnement adapté pour la méthode k-fold, par exemple StratifiedKFold dans Scikit-learn.
Un échantillonnage stratifié pour la méthode shuffle and split, par exemple StratifiedShuffleSplit dans Scikit-learn.
La méthode leave one out (LOO) peut être employée telle quelle.
Pour les problèmes dans lesquels les observations ne sont pas indépendantes :
Dans le cas des séries temporelles, les observations successives sont corrélées. Le découpage doit alors être fait par séquences (permettant de conserver un historique local) sur les observations ordonnées et non après shuffle sur les observations individuelles.
Dans le cas des données groupées, à l’intérieur d’un même groupe les observations ne sont pas indépendantes. Les données de test doivent alors provenir de groupes différents de ceux dont sont issues les données d’apprentissage.
L’évaluation de base d’un modèle de classement (ou modèle discriminant) sur un ensemble de données \(\mathcal{E}\) est réalisée à travers le taux de mauvais classement des données de l’ensemble de test, qui correspond à l’erreur moyenne (sur les données de test) obtenue en utilisant la perte 0-1. Une telle évaluation est toutefois réductrice car la perte 0-1 fait l’hypothèse d’un coût de mauvais classement symétrique : le coût est égal à 1 quelle que soit le sens de l’erreur d’affectation. Or, dans de nombreux cas les erreurs d’affectation ne sont pas équivalentes. Par exemple, ne pas détecter la maladie grave d’un patient à partir de mesures cliniques est dramatique (observation de la classe 1 affectée par erreur à la classe 0), alors que signaler une maladie grave pour un patient qui n’en est pas atteint oblige simplement à effectuer un ou plusieurs examens complémentaires (observation de la classe 0 affectée par erreur à la classe 1). Un autre exemple : pour un cargo, la non détection d’un autre navire par le radar peut mener à une collision, alors qu’une fausse alerte provoque seulement un ralentissement temporaire. Bien entendu, si cette asymétrie du coût peut être quantifiée alors elle peut être prise en compte dès le départ, dans la construction du modèle décisionnel. Il est toutefois utile de pouvoir comparer plusieurs modèles discriminants sans fixer a priori un « degré » d’asymétrie.
Les courbes ROC (initiales provenant de receiver operating characteristic, ou « caractéristique de fonctionnement du récepteur » en traduction littérale) répondent à cet objectif. On considère un problème de discrimination dans lequel une des classes est la « classe d’intérêt » et un modèle de classement (ou discriminant) appris à partir des données est vu comme le « détecteur » de la classe d’intérêt.
Un modèle discriminant est en général décrit par un vecteur de paramètres (par ex. \(\mathbf{w}\), qui indique l’orientation de l’hyperplan de séparation pour un modèle linéaire) et un seuil de détection (\(b\), la position de l’hyperplan pour le modèle linéaire). Le principe d’une courbe ROC est de balayer la totalité du domaine utile de variation de ce seuil et d’examiner, pour chaque valeur du seuil, les résultats obtenus sur les données d’évaluation, représentés par la matrice de confusion :
Classe présente
Classe absente
Classe détectée
Vrai Positif
Faux Positif
Classe non détectée
Faux Négatif
Vrai Négatif
Chaque donnée de \(\mathcal{E}\) peut appartenir à la classe d’intérêt (Classe présente) ou à une autre classe (Classe [d’intérêt] absente) ; cette information est supposée connue pour toutes les données de \(\mathcal{E}\) (« vérité terrain »). Chaque donnée peut être affectée par le modèle à la classe d’intérêt (Classe détectée) ou à une autre classe (Classe non détectée). Si une donnée fait partie de la classe d’intérêt mais est affectée par le modèle à une autre classe, on parle d’un « faux négatif ». Si une donnée est affectée par le modèle à la classe d’intérêt alors qu’elle fait partie d’une autre classe, on parle d’un « faux positif ».
A partir du contenu d’une matrice de confusion, on définit les mesures suivantes :
La sensibilité, ou le taux de vrais positifs, mesure la capacité du modèle à détecter les vrais positifs. Si tous les vrais positifs sont détectés (s’il n’y a pas de faux négatifs, c’est à dire des positifs non détectés comme positifs), alors la sensibilité est égale à 1.
La 1 - spécificité, ou le taux de faux positifs, mesure la capacité du modèle à détecter seulement les vrais positifs. Si aucun négatif n’est détecté comme positif (s’il n’y a pas de faux positifs), alors la 1 - spécificité est égale à 0.
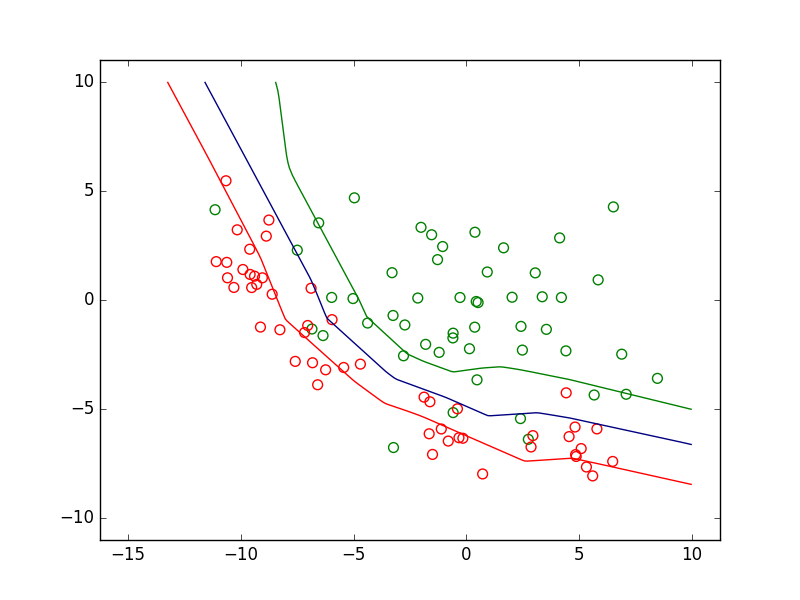
Dans la figure suivante sont représentées des données de \(\mathbb{R}^2\). Celles de la classe d’intérêt sont en vert et celles des autres classes en rouge. Considérons trois modèles discriminants qui sont des PMC avec \(\alpha = 1\) correspondant à trois valeurs différentes du seuil. Chaque modèle discriminant est une frontière de séparation non linéaire. Dans cet exemple, la classe d’intérêt n’est jamais parfaitement séparée des autres classes, quelle que soit la position de la frontière.
Fig. 27 Frontières de discrimination pour 3 valeurs du seuil de détection#
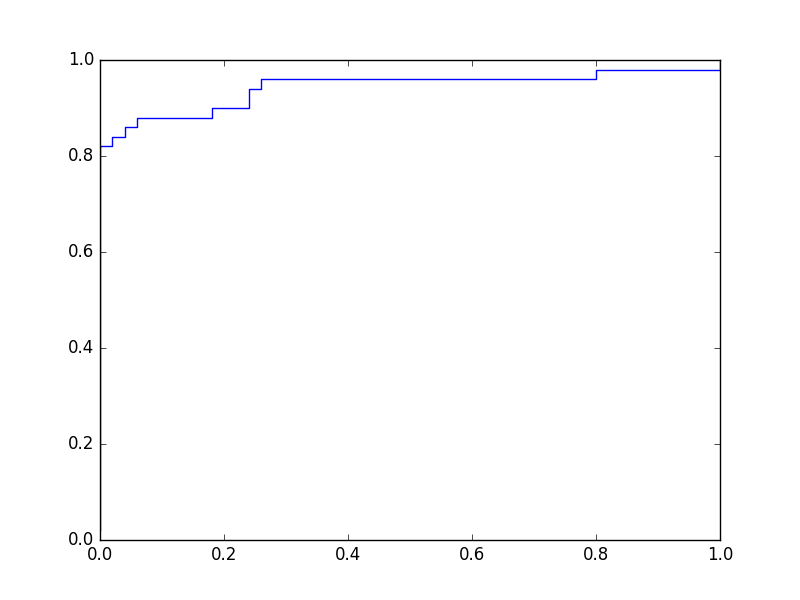
Une courbe ROC montre le taux de vrais positifs (en ordonnée) en fonction du taux de faux positifs (en abscisse), la variable étant le seuil. Pour le problème illustré dans la figure précédente, en explorant la totalité du domaine de variation utile du seuil et en enregistrant à chaque fois la sensibilité (ou taux de vrais positifs) et 1 - spécificité (ou taux de faux positifs), on obtient le graphique suivant :
Fig. 28 Courbe ROC associée à l’exemple de la Fig. 27#
Lorsque la frontière est positionnée très haut dans la Fig. 27, toutes le données sont du côté « négatif ». Aucun vrai positif n’est détecté et aucun négatif n’est détecté comme positif. La sensibilité est donc nulle et la 1 - spécificité est nulle également.
Au fur et à mesure que la frontière se déplace vers le bas et vers la gauche dans la Fig. 27, de plus en plus de positifs sont détectés mais aucun négatif n’est encore détecté comme positif. La sensibilité augmente et la 1 - spécificité reste nulle. A partir d’une valeur du seuil, des négatifs commencent à être détectés comme positifs. La 1 - spécificité commence donc à augmenter. Si la frontière continue son déplacement vers le bas et la gauche, à partir d’une autre valeur du seuil il n’y a plus aucun positif du mauvais côté de la frontière, tous sont détectés comme des positifs. La sensibilité atteint donc 1. Noter que dans l’exemple de la Fig. 27 un des points verts est très éloigné de sa classe, ce fait que la sensibilité n’atteint pas 1 avant que la 1 - spécificité ait atteint 1, comme on peut le voir sur la courbe ROC de la Fig. 28. En revanche, comme de plus en plus de négatifs sont détectés comme positifs, la 1 - spécificité continue à augmenter.
Lorsque la frontière est suffisamment en bas à gauche dans la Fig. 27, toutes le données sont du côté « positif ». Tous les positifs sont détectés et, en plus, tous les négatifs sont détectés comme positifs. La sensibilité est donc égale à 1 mais la 1 - spécificité est égale à 1 aussi. Noter que ce cas n’est pas illustré sur la Fig. 28 car on s’arrête lorsque la 1 - spécificité atteint 1 même si la sensibilité est encore inférieure à 1.
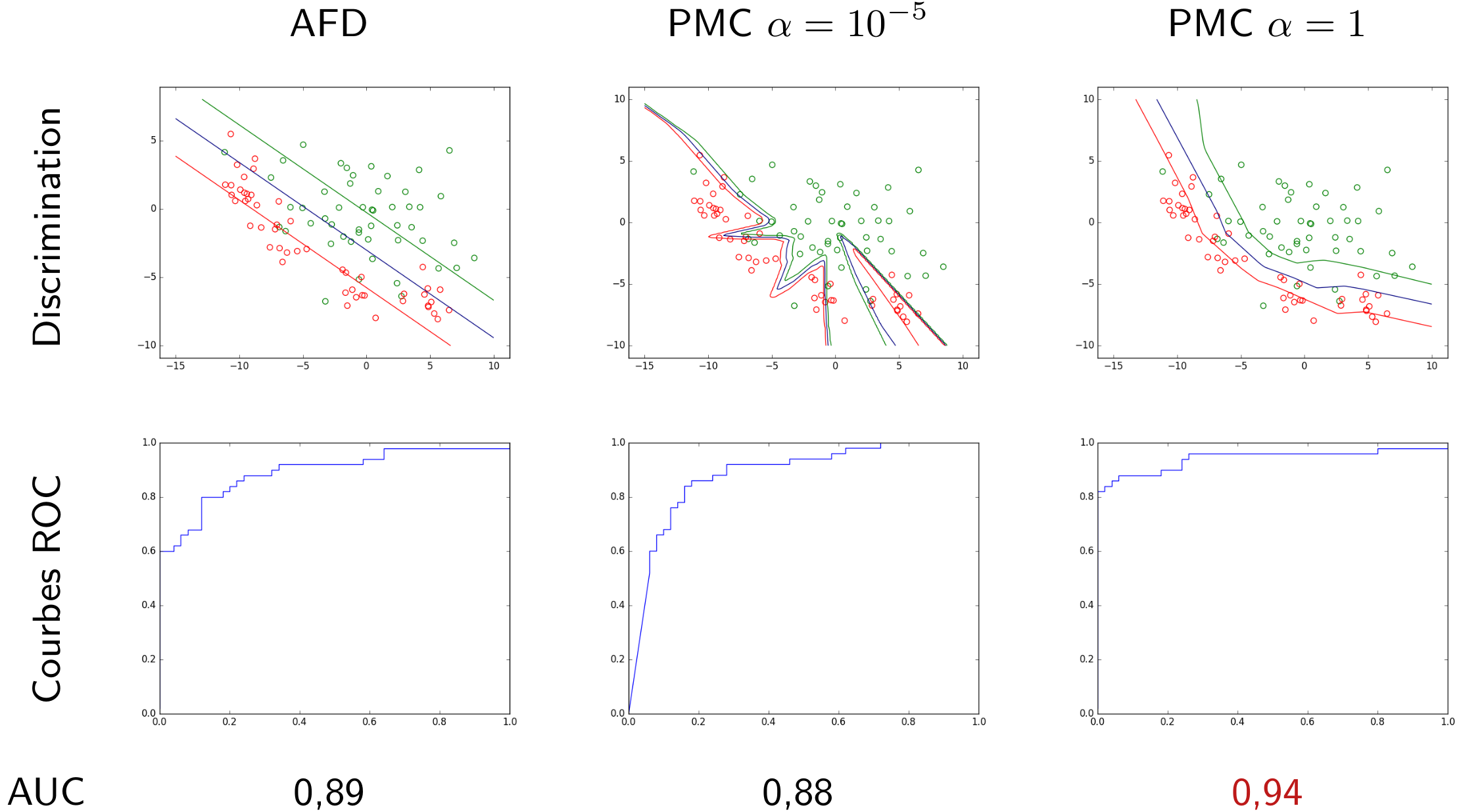
Peut-on comparer ces modèles à partir de leurs courbes ROC, pour constater éventuellement qu’un est plus intéressant (ou moins intéressant) que les autres sur la totalité du domaine de variation du seuil ? Comparons les résultats avec ceux de deux autres modèles, un modèle linéaire (obtenu par AFD) et un autre qui est un PMC avec \(\alpha = 10^{-5}\). La figure suivante montre les frontières de discrimination obtenues par chaque modèle pour trois valeurs différentes du seuil, ainsi que les courbes ROC correspondantes :
Fig. 29 Comparaison de modèles à travers leurs courbes ROC et les aires sous la courbe#
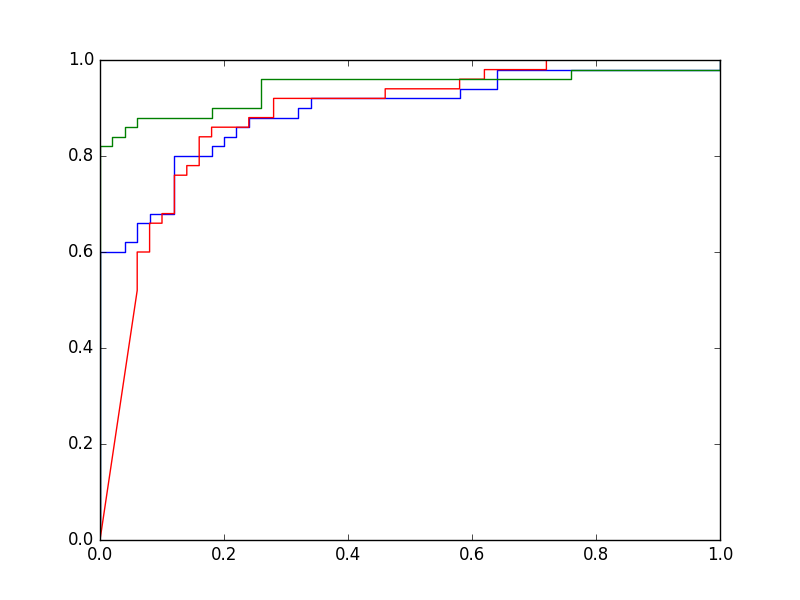
Les trois courbes ROC regroupées sur un même graphique sont montrées dans la figure suivante. La courbe ROC du PMC \(\alpha = 1\) se situe presque systématiquement au-dessus des deux autres, en revanche les courbes ROC des deux autres modèles se croisent plusieurs fois. Une mesure couramment employée pour comparer deux modèles sur la totalité du domaine de variation utile du seuil est l”aire sous la courbe ROC, notée en général AUC (area under curve). Plus sa valeur est élevée, plus le modèle est intéressant (performant). Si deux modèles ont des valeurs AUC très proches, le choix doit être fait à partir d’autres critères, par exemple en comparant les sensibilités à spécificité fixée.
Fig. 30 Courbes ROC : modèle linéaire en bleu, PMC \(\alpha = 10^{-5}\) en rouge, PMC \(\alpha = 1\) en vert#
Le traitement d’un problème de modélisation décisionnelle à partir d’un ensemble de données \(\mathcal{D}_N\) passe en général par l’évaluation de plusieurs « procédures » de modélisation différentes, chacune définie par une famille de modèles \(\mathcal{F}\), des paramètres spécifiques à la famille choisie (par ex. l’architecture pour un PMC, le type et la variance du noyau pour une SVM), un critère de régularisation \(G(f(\mathbf{w}))\), une pondération pour le terme de régularisation, etc.
Les paramètres numériques dont dépend une procédure de modélisation sont en général appelés « hyperparamètres », bien que ce terme ait été employé à l’origine pour les paramètres des distributions a priori dans les statistiques bayesiennes. Par exemple, pour les SVM à noyau RBF sont considérés hyperparamètres la variance du noyau RBF et la valeur de la constante \(C\) de régularisation. Parfois, le terme « hyperparamètres » est étendu aux variables nominales définissant le famille de modèles \(\mathcal{F}\), comme par exemple le type de noyau employé pour des SVM.
Le différentes « procédures » de modélisation sont ensuite comparées à travers leurs scores de validation croisée afin de retenir celle pour laquelle le risque espéré (l’erreur de généralisation) présente la valeur la plus faible. La « procédure » ainsi choisie est enfin employée pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). Une fois trouvé le « meilleur » modèle (par la procédure de modélisation retenue), son risque espéré doit être estimé sur des données de qui n’ont servi ni à la recherche des paramètres, ni à celle des hyperparamètres !
Pour trouver le meilleur modèle décisionnel il est nécessaire de comparer plusieurs « procédures » de modélisation différentes. Chaque « procédure » de modélisation est définie par une valeur spécifique pour chaque hyperparamètre. Une fois fixées les valeurs des hyperparamètres (par ex. de \(C\) pour les SVM linéaires), l’algorithme d’optimisation (par ex. la descente de sous-gradient stochastique) opère pour déterminer le modèle (par ex. \(\mathbf{w}^*\) et \(b^*\) pour une SVM linéaire).
Les meilleures valeurs des hyperparamètres sont identifiées de la manière suivante :
Définition d’ensembles de valeurs pour les éventuels hyperparamètres nominaux (par ex. architectures PMC, critères de régularisation, types de noyaux pour les SVM).
Définition d’intervalles de variation pour les hyperparamètres numériques et d’une procédure d’exploration de cet espace.
Exploration de l’espace des hyperparamètres suivant la procédure choisie. Pour chaque tuple de valeurs (une valeur pour chaque hyperparamètre), apprentissage et évaluation des modèles résultants par application de la validation croisée.
Comparaison des résultats de validation croisée, sélection des valeurs des hyperparamètres qui mènent aux meilleures performances (à l’erreur de généralisation estimée la plus faible).
Une procédure d’exploration simple est la recherche systématique dans une grille (grid search) : si on dispose de \(m\) hyperparamètres numériques, on définit pour chaque hyperparamètre un intervalle et un pas de variation, on obtient ainsi une grille (de dimension \(m\)) dont les nœuds correspondent aux valeurs à tester. Pour chaque nœud de la grille (tuple de \(m\) valeurs), la validation croisée est appliquée afin d’obtenir une estimation de l’erreur de généralisation des modèles correspondants. Bien entendu, tous les nœuds de la grille peuvent être explorés en parallèle si une plate-forme adéquate est disponible.
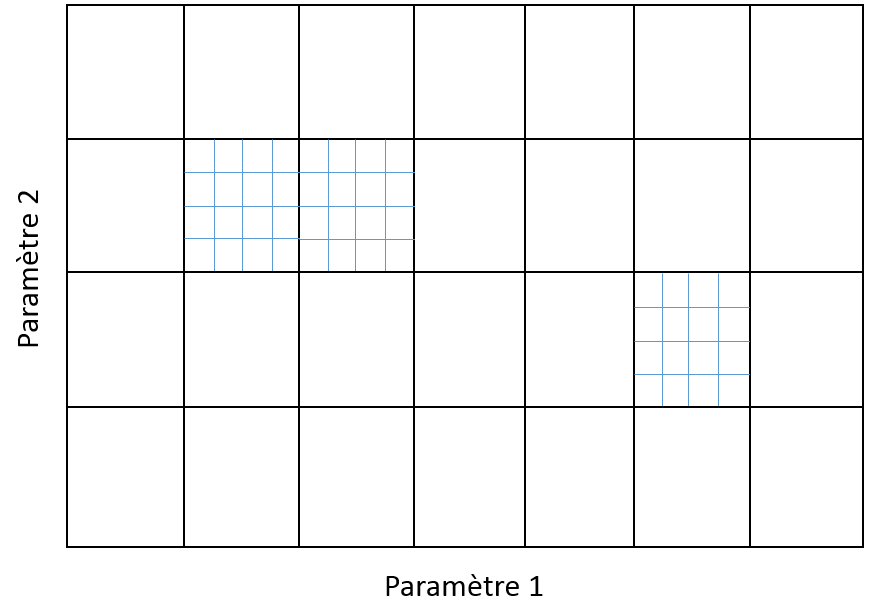
Avec une grille trop fine, le nombre de modèles à explorer peut être excessif. Avec une grille trop grossière, l’exploration de l’espace des paramètres peut être trop grossière et ne pas permettre de trouver les meilleurs modèles. Une solution souvent adoptée est celle d’une grille qui a plusieurs niveaux de « finesse », comme dans la figure suivante. Dans un tel cas, la recherche est hiérarchique : sont d’abord explorées les valeurs correspondant à la grille grossière, ensuite des grilles plus fines sont définies dans les intervalles de la grille grossière où les meilleurs résultats ont été trouvés, et sont explorées enfin les valeurs issues de ces grilles plus fines. Cette exploration hiérarchique permet d’obtenir une exploration plus fine à moindre coût qu’une recherche exhaustive au niveau le plus fin. Le procédé peut être répété à plusieurs niveaux. A chaque niveau, les nœuds de la grille peuvent être explorés en parallèle.
Fig. 31 Grille à deux niveaux pour recherche hiérarchique#
Il est important de noter que pour des méthodes d’apprentissage spécifiques il est parfois possible de définir des procédures d’exploration plus efficaces.
Les valeurs des hyperparamètres qui mènent à l’erreur de généralisation estimée la plus faible sont retenues et employées pour apprendre un modèle sur la totalité des données de \(\mathcal{D}_N\). C’est ce modèle qui est retourné par la procédure de recherche en grille.
Enfin, pour estimer le risque espéré (l’erreur de généralisation) du modèle retenu in fine il faut employer des données qui n’ont été utilisées ni pour l’apprentissage du modèle (pour l’algorithme d’optimisation des paramètres), ni pour la recherche des meilleures valeurs des hyperparamètres !
Ceci permettra d’éviter des estimations trop optimistes (valeur estimée trop basse pour le risque espéré).
Lorsque des connaissances a priori permettent de privilégier certains intervalles de variation pour les hyperparamètres, la recherche aléatoire à partir de valeurs générées en conformité avec ces connaissances présente une meilleure efficacité qu’une grid search non hiérarchique.
Concrètement, pour chaque hyperparamètre une loi de tirage est définie, ensuite des tuples de valeurs (une valeur pour chaque hyperparamètre) sont obtenues et un modèle est appris avec ce tuple de valeurs. Les échantillons sont générés en considérant en général les hyperparamètres comme étant indépendants. Un intérêt particulier de cette approche est que le coût peut être maîtrisé en fixant le nombre d’échantillons à générer.
Les modalités d’échantillonnage dépendent de la nature des hyperparamètres considérés :
pour les hyperparamètres numériques à valeurs continues (par ex. la pondération \(\alpha\) de la régularisation) on précise la loi d’échantillonnage, par ex. loi gamma de paramètres donnés ;
pour les hyperparamètres numériques à valeurs discrètes (par ex. nombre de neurones cachés) on précise la loi d’échantillonnage, par ex. loi uniforme sur un intervalle donné ;
pour les hyperparamètres variables nominales, on précise la liste des valeurs (modalités) possibles et on considère une loi uniforme sur ces valeurs.
Évaluation et sélection de modèles : que faut-il retenir ?#
Le risque espéré (l’erreur de généralisation) doit être estimé sur des données non utilisées pour l’apprentissage.
La validation croisée permet une estimation de plus faible variance qu’un seul découpage apprentissage | test, ainsi qu’une meilleure utilisation des données disponibles.
Le courbes ROC facilitent une comparaison plus globale de modèles de classement (ou de discrimination).
Pour obtenir les meilleures valeurs pour les hyperparamètres on emploie une recherche systématique ou aléatoire, la comparaison des modèles étant réalisée par validation croisée.
Si la validation croisée est employée pour sélectionner le meilleur modèle, l’estimation du risque espéré du modèle retenu doit être faite sur des données non encore utilisées.
[BBL04]
O. Bousquet, S. Boucheron, and G. Lugosi. Introduction to Statistical Learning Theory, pages 169–207. Volume Lecture Notes in Artificial Intelligence 3176. Springer, Heidelberg, Germany, 2004.
[CScholkopfZ06]
Olivier Chapelle, Bernhard Schölkopf, and Alexander Zien, editors. Semi-Supervised Learning. MIT Press, Cambridge, MA, 2006. URL: http://www.kyb.tuebingen.mpg.de/ssl-book.
Bernhard Schölkopf and Alexander Smola. Learning with Kernels. MIT Press, 2002.
[YRCF17]
Lixuan Yang, Helena Rodriguez, Michel Crucianu, and Marin Ferecatu. Fully convolutional network with superpixel parsing for fashion web image segmentation. In Proc. 23rd Intl. Conf. MultiMedia Modeling, Reykjavik, Iceland, 139–151. 2017.